

Mutations in SKI in Shprintzen-Goldberg syndrome lead to attenuated TGF-β responses through SKI stabilization
- PMID: 33416497
- PMCID: PMC7834018
- DOI: 10.7554/eLife.63545
Mutations in SKI in Shprintzen-Goldberg syndrome lead to attenuated TGF-β responses through SKI stabilization
Abstract
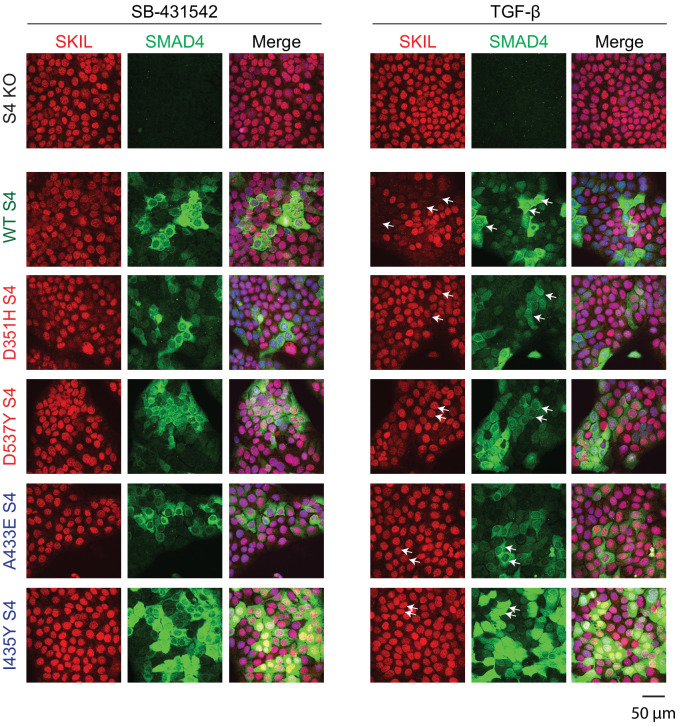
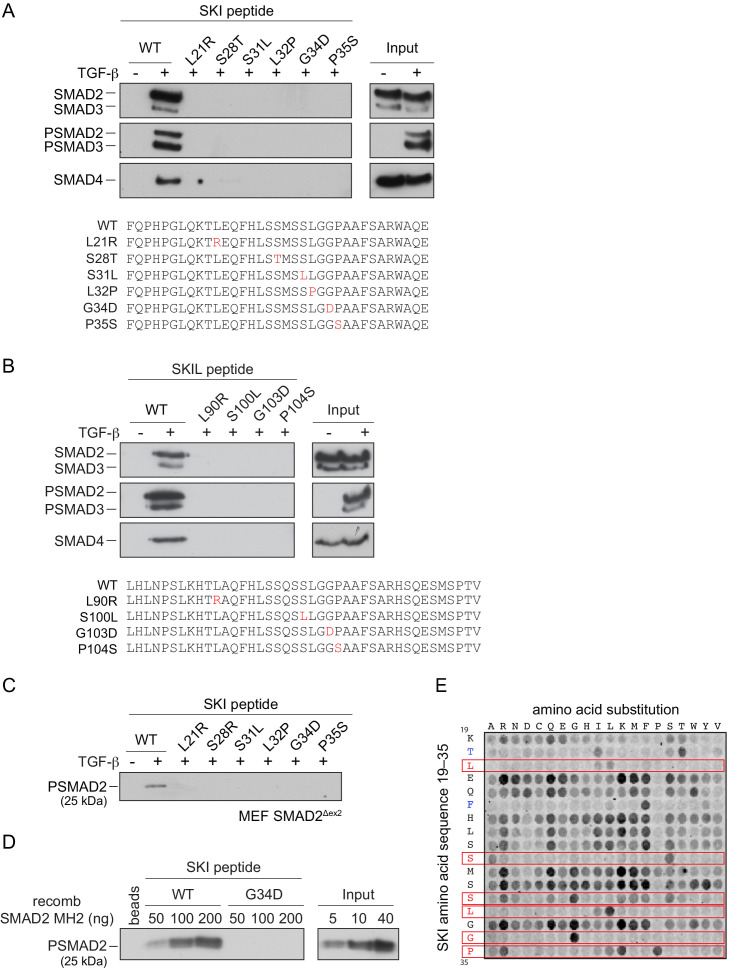
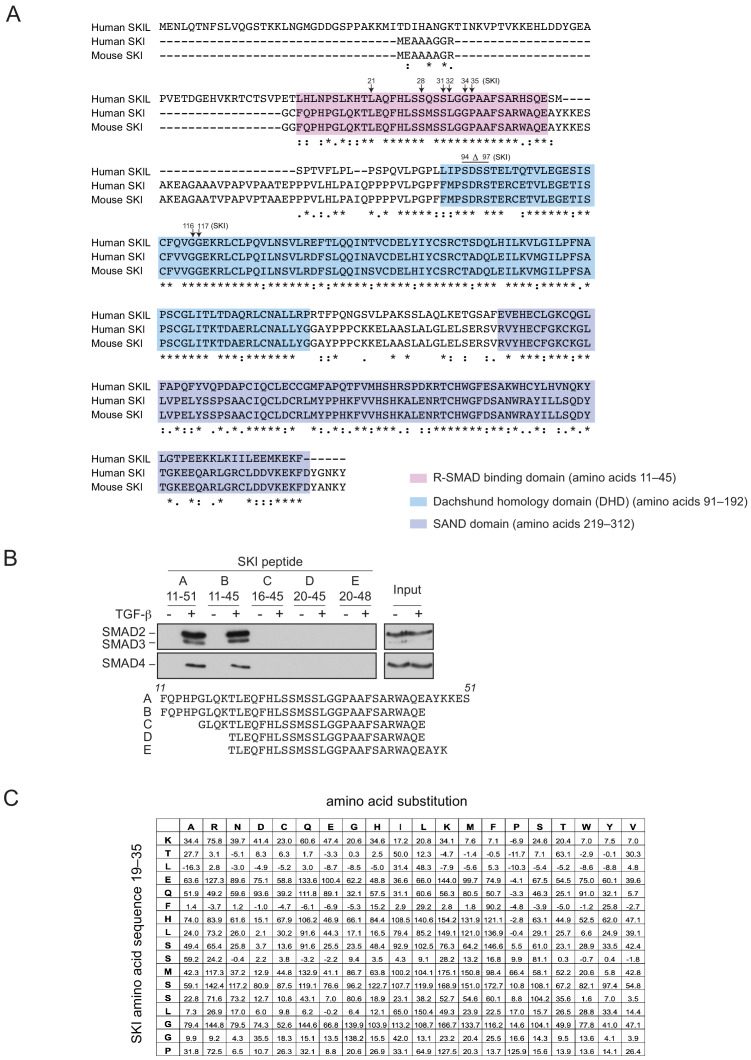
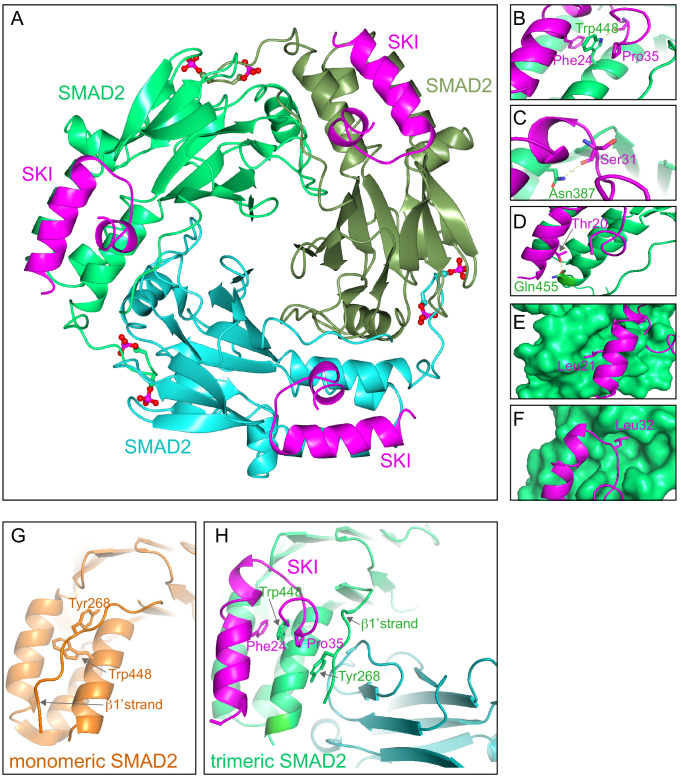
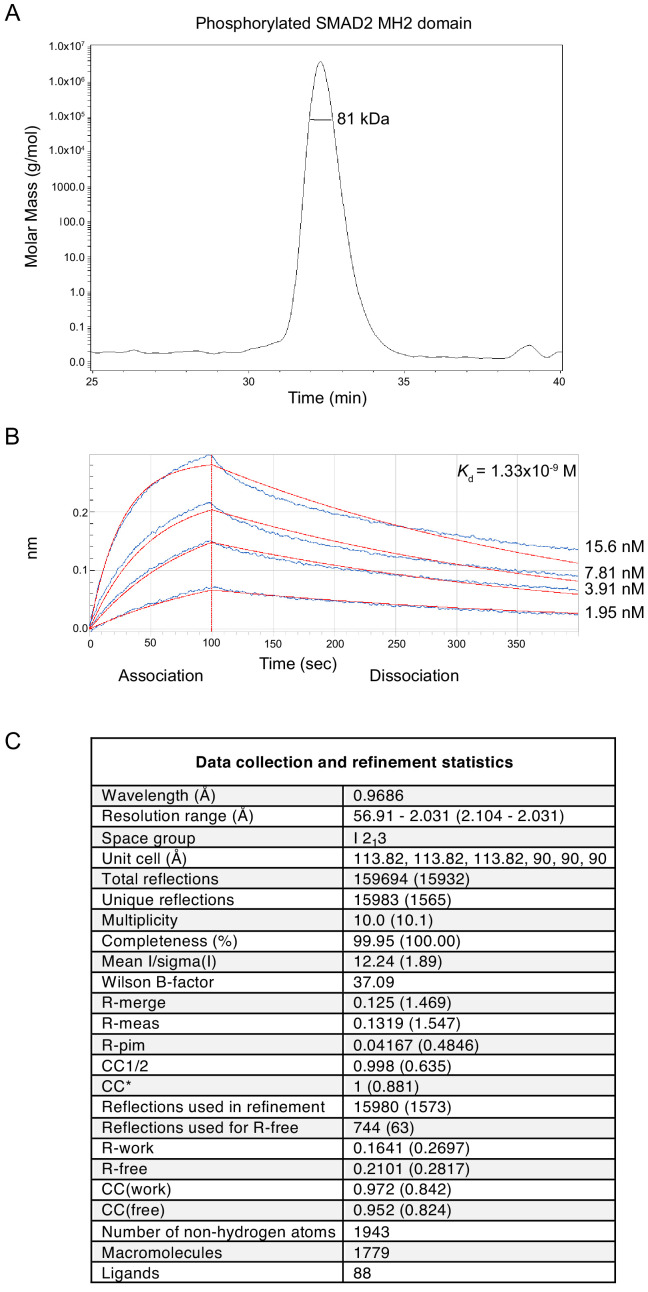
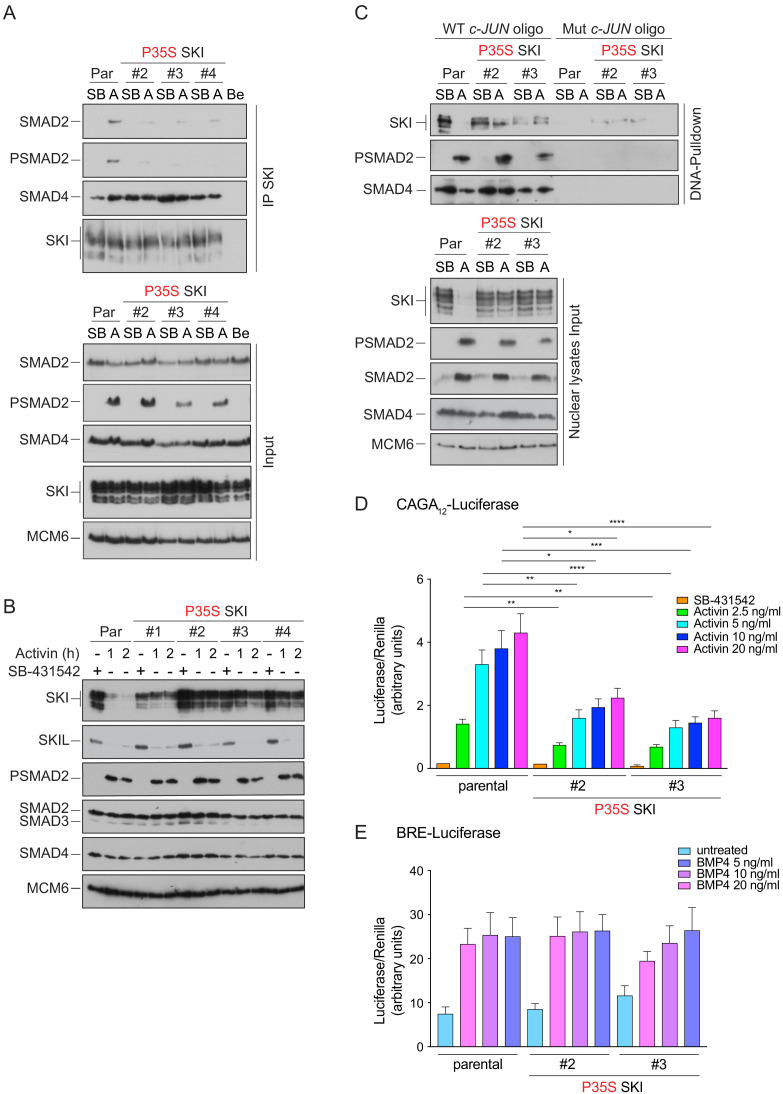
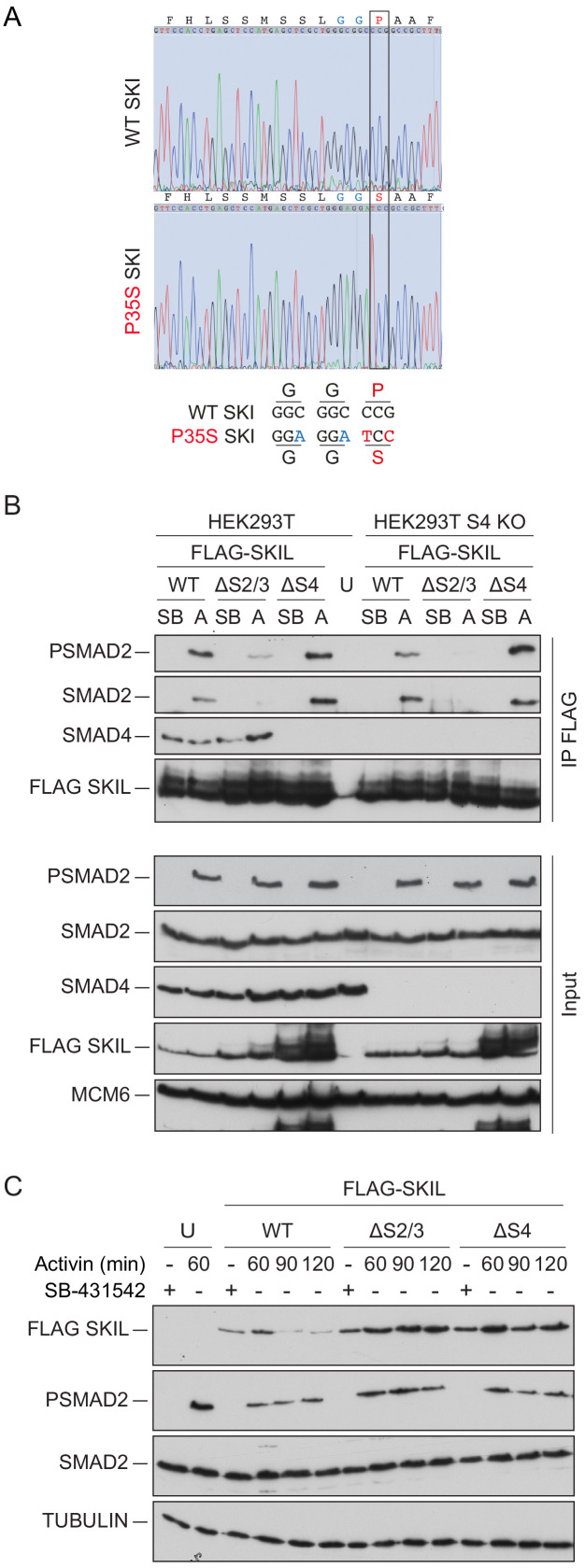
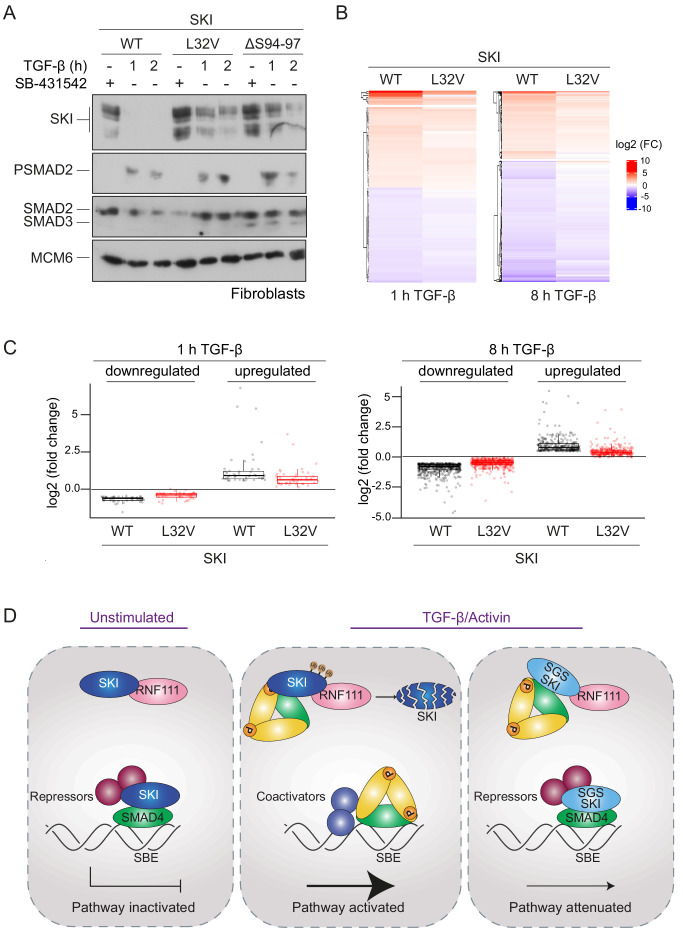
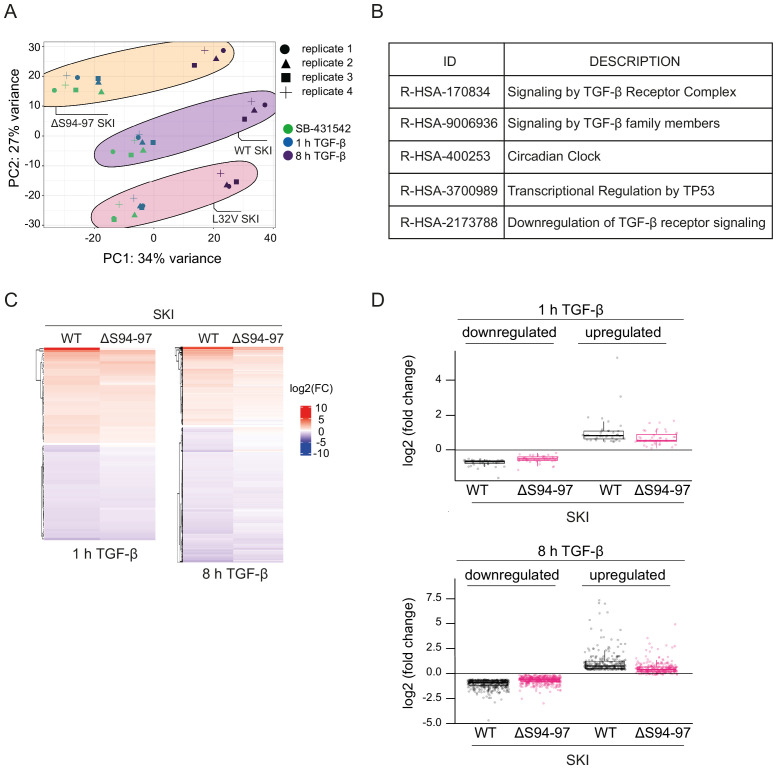
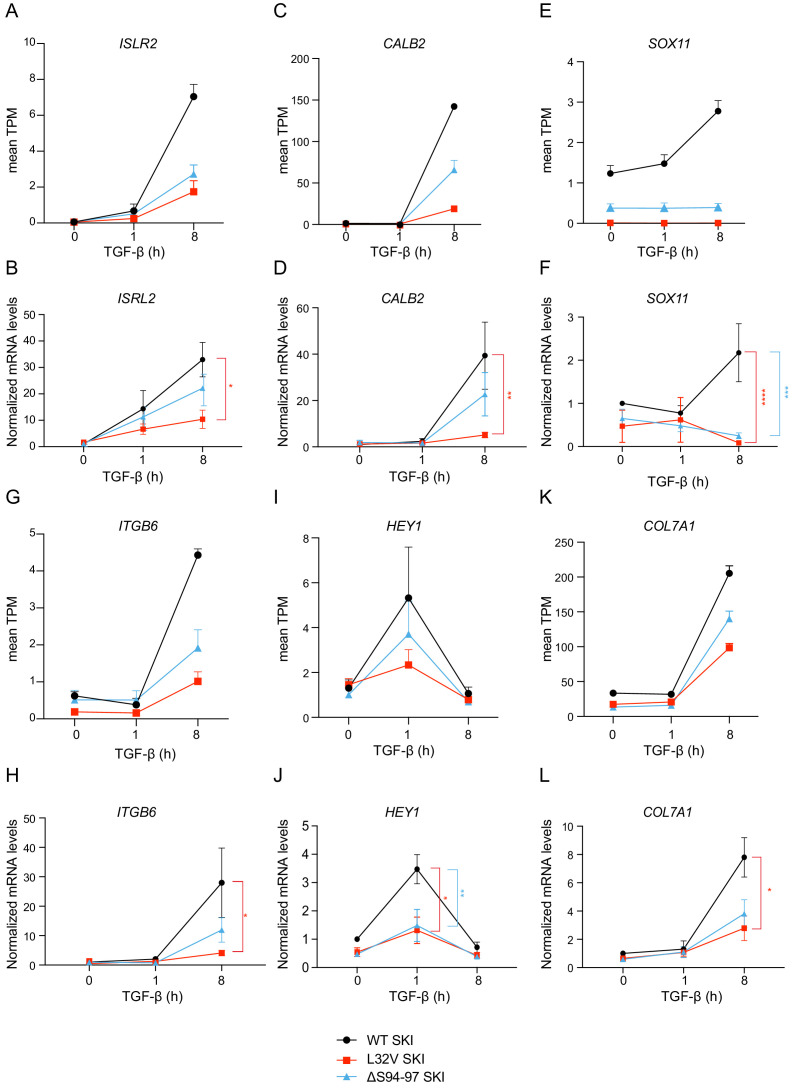
Shprintzen-Goldberg syndrome (SGS) is a multisystemic connective tissue disorder, with considerable clinical overlap with Marfan and Loeys-Dietz syndromes. These syndromes have commonly been associated with enhanced TGF-β signaling. In SGS patients, heterozygous point mutations have been mapped to the transcriptional co-repressor SKI, which is a negative regulator of TGF-β signaling that is rapidly degraded upon ligand stimulation. The molecular consequences of these mutations, however, are not understood. Here we use a combination of structural biology, genome editing, and biochemistry to show that SGS mutations in SKI abolish its binding to phosphorylated SMAD2 and SMAD3. This results in stabilization of SKI and consequently attenuation of TGF-β responses, both in knockin cells expressing an SGS mutation and in fibroblasts from SGS patients. Thus, we reveal that SGS is associated with an attenuation of TGF-β-induced transcriptional responses, and not enhancement, which has important implications for other Marfan-related syndromes.
Keywords: Activin; Marfan syndrome; SKI; SMAD; Shprintzen-Goldberg syndrome; TGF-β; biochemistry; chemical biology; chromosomes; gene expression; human.
© 2021, Gori et al.
Conflict of interest statement
IG, RG, AP, SS, RR, RO, VC, LF, DJ, SK, CH No competing interests declared
Figures
Similar articles
-
Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm.Nat Genet. 2012 Nov;44(11):1249-54. doi: 10.1038/ng.2421. Epub 2012 Sep 30. Nat Genet. 2012. PMID: 23023332 Free PMC article.
-
The SMAD-binding domain of SKI: a hotspot for de novo mutations causing Shprintzen-Goldberg syndrome.Eur J Hum Genet. 2015 Feb;23(2):224-8. doi: 10.1038/ejhg.2014.61. Epub 2014 Apr 16. Eur J Hum Genet. 2015. PMID: 24736733 Free PMC article.
-
Shprintzen - Goldberg syndrome without intellectual disability: A clinical report and review of literature.Eur J Med Genet. 2025 Feb;73:104985. doi: 10.1016/j.ejmg.2024.104985. Epub 2024 Dec 3. Eur J Med Genet. 2025. PMID: 39638120 Review.
-
De novo exon 1 missense mutations of SKI and Shprintzen-Goldberg syndrome: two new cases and a clinical review.Am J Med Genet A. 2014 Mar;164A(3):676-84. doi: 10.1002/ajmg.a.36340. Epub 2013 Dec 19. Am J Med Genet A. 2014. PMID: 24357594 Review.
-
In-frame mutations in exon 1 of SKI cause dominant Shprintzen-Goldberg syndrome.Am J Hum Genet. 2012 Nov 2;91(5):950-7. doi: 10.1016/j.ajhg.2012.10.002. Epub 2012 Oct 25. Am J Hum Genet. 2012. PMID: 23103230 Free PMC article.
Cited by
-
Meticulous and Early Understanding of Congenital Cranial Defects Can Save Lives.Children (Basel). 2023 Jul 18;10(7):1240. doi: 10.3390/children10071240. Children (Basel). 2023. PMID: 37508737 Free PMC article.
-
Disruptor: Computational identification of oncogenic mutants disrupting protein-protein and protein-DNA interactions.Commun Biol. 2023 Jul 13;6(1):720. doi: 10.1038/s42003-023-05089-2. Commun Biol. 2023. PMID: 37443295 Free PMC article.
-
Insights on the Pathogenesis of Aneurysm through the Study of Hereditary Aortopathies.Genes (Basel). 2021 Jan 27;12(2):183. doi: 10.3390/genes12020183. Genes (Basel). 2021. PMID: 33514025 Free PMC article. Review.
-
Role of elastic fiber degradation in disease pathogenesis.Biochim Biophys Acta Mol Basis Dis. 2023 Jun;1869(5):166706. doi: 10.1016/j.bbadis.2023.166706. Epub 2023 Mar 29. Biochim Biophys Acta Mol Basis Dis. 2023. PMID: 37001705 Free PMC article. Review.
-
Context-dependent TGFβ family signalling in cell fate regulation.Nat Rev Mol Cell Biol. 2023 Dec;24(12):876-894. doi: 10.1038/s41580-023-00638-3. Epub 2023 Aug 18. Nat Rev Mol Cell Biol. 2023. PMID: 37596501 Review.
References
-
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallographica Section D Biological Crystallography. 2012;68:352–367. doi: 10.1107/S0907444912001308. - DOI - PMC - PubMed
-
- Angelov SN, Hu JH, Wei H, Airhart N, Shi M, Dichek DA. TGF-β (Transforming growth Factor-β) Signaling protects the thoracic and abdominal aorta from angiotensin II-Induced pathology by distinct mechanisms. Arteriosclerosis, Thrombosis, and Vascular Biology. 2017;37:2102–2113. doi: 10.1161/ATVBAHA.117.309401. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
