

Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK
- PMID: 33419936
- PMCID: PMC7877493
- DOI: 10.1126/science.abf2946
Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK
Abstract
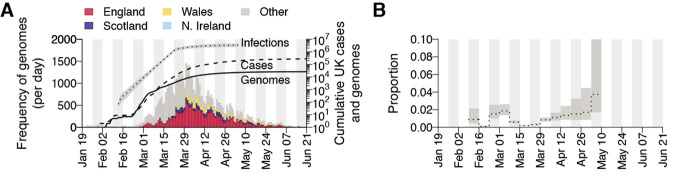
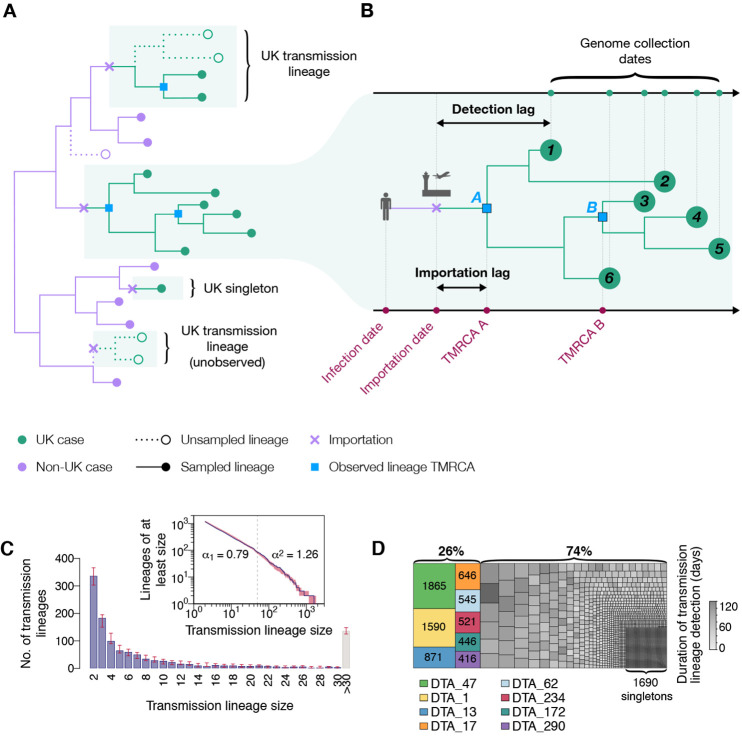
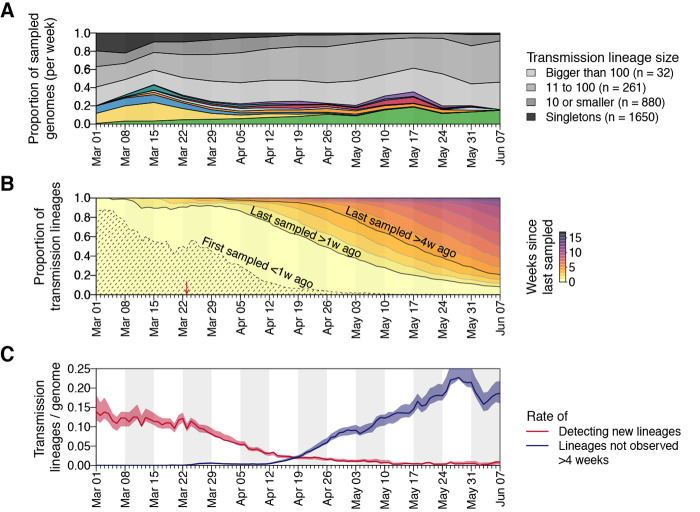
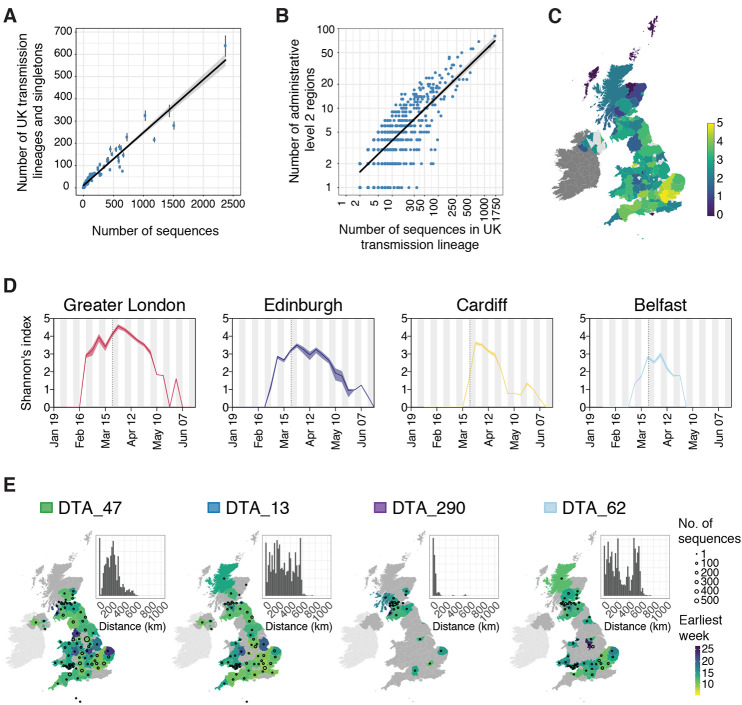
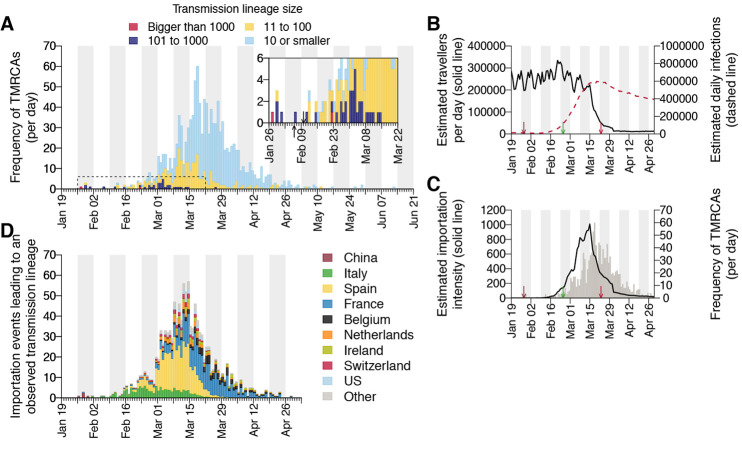
The United Kingdom's COVID-19 epidemic during early 2020 was one of world's largest and was unusually well represented by virus genomic sampling. We determined the fine-scale genetic lineage structure of this epidemic through analysis of 50,887 severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genomes, including 26,181 from the UK sampled throughout the country's first wave of infection. Using large-scale phylogenetic analyses combined with epidemiological and travel data, we quantified the size, spatiotemporal origins, and persistence of genetically distinct UK transmission lineages. Rapid fluctuations in virus importation rates resulted in >1000 lineages; those introduced prior to national lockdown tended to be larger and more dispersed. Lineage importation and regional lineage diversity declined after lockdown, whereas lineage elimination was size-dependent. We discuss the implications of our genetic perspective on transmission dynamics for COVID-19 epidemiology and control.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures
Comment in
-
Tracking the UK SARS-CoV-2 outbreak.Science. 2021 Feb 12;371(6530):680-681. doi: 10.1126/science.abg2297. Science. 2021. PMID: 33574202 No abstract available.
References
-
- Centers for Disease Control and Prevention , Severe acute respiratory syndrome - Singapore, 2003. MMWR Morb. Mortal. Wkly. Rep. 52, 405–411 (2003). - PubMed
-
- Faye O., Boëlle P. Y., Heleze E., Faye O., Loucoubar C., Magassouba N., Soropogui B., Keita S., Gakou T., Bah H. I., Koivogui L., Sall A. A., Cauchemez S., Chains of transmission and control of Ebola virus disease in Conakry, Guinea, in 2014: An observational study. Lancet Infect. Dis. 15, 320–326 (2015). 10.1016/S1473-3099(14)71075-8 - DOI - PMC - PubMed
-
- Bahl J., Nelson M. I., Chan K. H., Chen R., Vijaykrishna D., Halpin R. A., Stockwell T. B., Lin X., Wentworth D. E., Ghedin E., Guan Y., Peiris J. S. M., Riley S., Rambaut A., Holmes E. C., Smith G. J. D., Temporally structured metapopulation dynamics and persistence of influenza A H3N2 virus in humans. Proc. Natl. Acad. Sci. U.S.A. 108, 19359–19364 (2011). 10.1073/pnas.1109314108 - DOI - PMC - PubMed
-
- Baillie G. J., Galiano M., Agapow P. M., Myers R., Chiam R., Gall A., Palser A. L., Watson S. J., Hedge J., Underwood A., Platt S., McLean E., Pebody R. G., Rambaut A., Green J., Daniels R., Pybus O. G., Kellam P., Zambon M., Evolutionary dynamics of local pandemic H1N1/2009 influenza virus lineages revealed by whole-genome analysis. J. Virol. 86, 11–18 (2012). 10.1128/JVI.05347-11 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- MC_PC_19012/MRC_/Medical Research Council/United Kingdom
- MR/L015080/1/MRC_/Medical Research Council/United Kingdom
- MR/M501621/1/MRC_/Medical Research Council/United Kingdom
- MR/R015600/1/MRC_/Medical Research Council/United Kingdom
- MR/S019510/1/MRC_/Medical Research Council/United Kingdom
- BB/M010996/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- RP-PG-0514-20018/DH_/Department of Health/United Kingdom
- MR/T030062/1/MRC_/Medical Research Council/United Kingdom
- MC_PC_19027/MRC_/Medical Research Council/United Kingdom
- MR/J014370/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
