

An electron transfer competent structural ensemble of membrane-bound cytochrome P450 1A1 and cytochrome P450 oxidoreductase
- PMID: 33420418
- PMCID: PMC7794467
- DOI: 10.1038/s42003-020-01568-y
An electron transfer competent structural ensemble of membrane-bound cytochrome P450 1A1 and cytochrome P450 oxidoreductase
Abstract
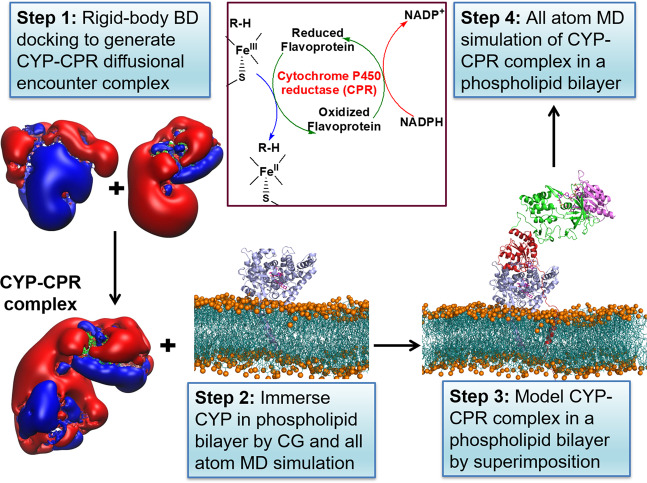
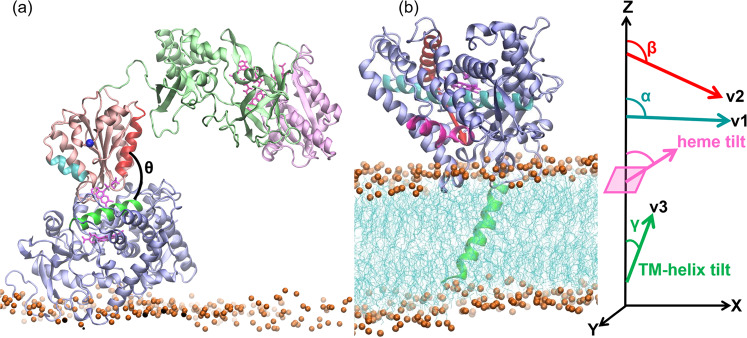
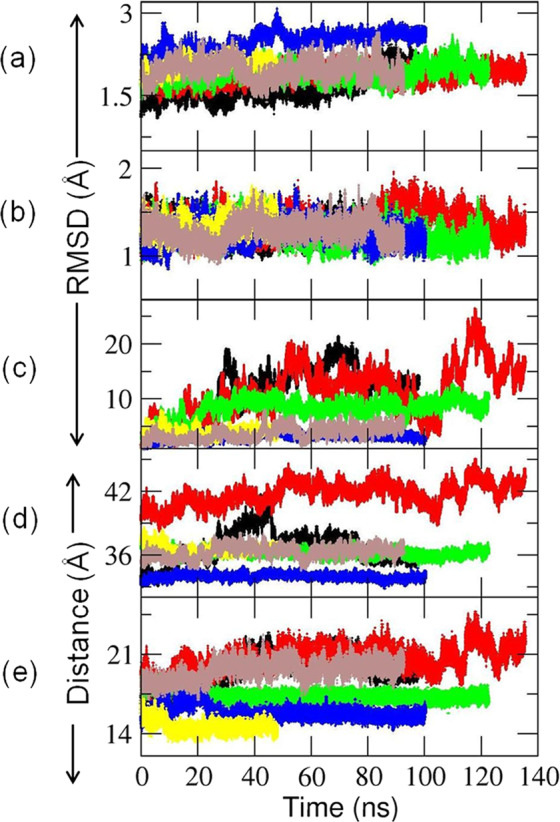
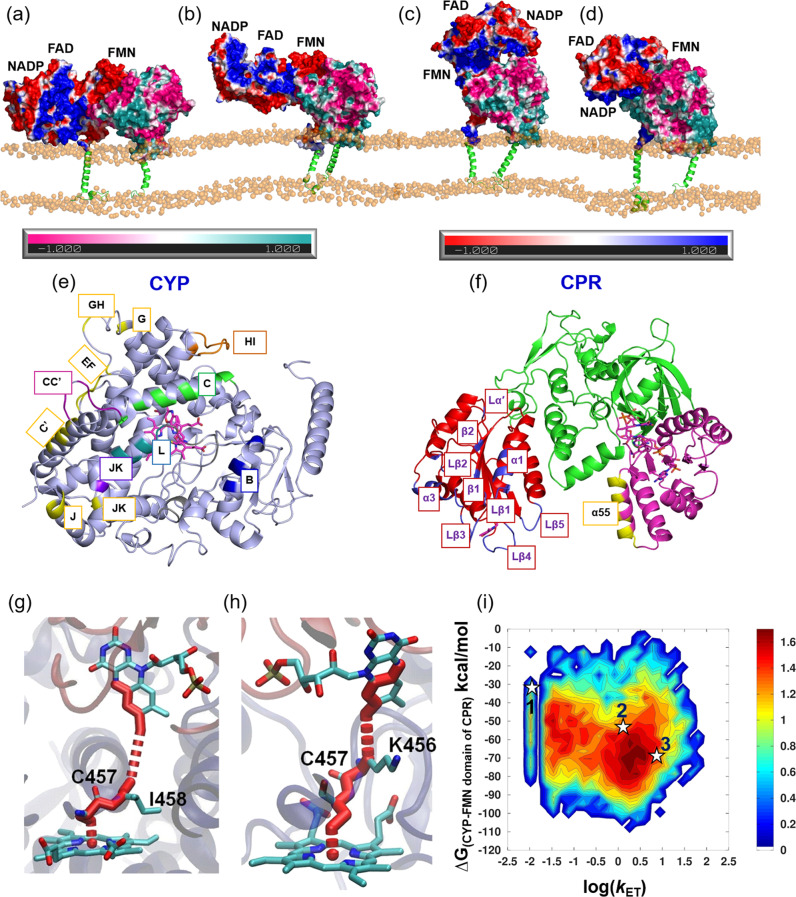
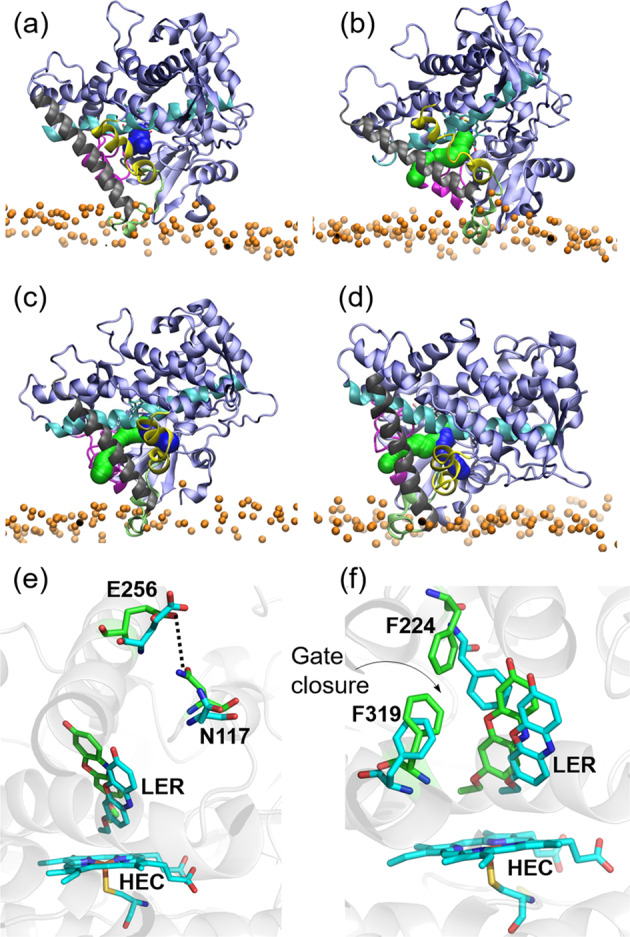
Cytochrome P450 (CYP) heme monooxygenases require two electrons for their catalytic cycle. For mammalian microsomal CYPs, key enzymes for xenobiotic metabolism and steroidogenesis and important drug targets and biocatalysts, the electrons are transferred by NADPH-cytochrome P450 oxidoreductase (CPR). No structure of a mammalian CYP-CPR complex has been solved experimentally, hindering understanding of the determinants of electron transfer (ET), which is often rate-limiting for CYP reactions. Here, we investigated the interactions between membrane-bound CYP 1A1, an antitumor drug target, and CPR by a multiresolution computational approach. We find that upon binding to CPR, the CYP 1A1 catalytic domain becomes less embedded in the membrane and reorients, indicating that CPR may affect ligand passage to the CYP active site. Despite the constraints imposed by membrane binding, we identify several arrangements of CPR around CYP 1A1 that are compatible with ET. In the complexes, the interactions of the CPR FMN domain with the proximal side of CYP 1A1 are supplemented by more transient interactions of the CPR NADP domain with the distal side of CYP 1A1. Computed ET rates and pathways agree well with available experimental data and suggest why the CYP-CPR ET rates are low compared to those of soluble bacterial CYPs.
Conflict of interest statement
The authors declare no competing interests.
Figures


Similar articles
-
The Role of the FMN-Domain of Human Cytochrome P450 Oxidoreductase in Its Promiscuous Interactions With Structurally Diverse Redox Partners.Front Pharmacol. 2020 Mar 18;11:299. doi: 10.3389/fphar.2020.00299. eCollection 2020. Front Pharmacol. 2020. PMID: 32256365 Free PMC article.
-
Probing the Role of the Hinge Segment of Cytochrome P450 Oxidoreductase in the Interaction with Cytochrome P450.Int J Mol Sci. 2018 Dec 6;19(12):3914. doi: 10.3390/ijms19123914. Int J Mol Sci. 2018. PMID: 30563285 Free PMC article.
-
Interaction Modes of Microsomal Cytochrome P450s with Its Reductase and the Role of Substrate Binding.Int J Mol Sci. 2020 Sep 11;21(18):6669. doi: 10.3390/ijms21186669. Int J Mol Sci. 2020. PMID: 32933097 Free PMC article.
-
Conformational changes of the NADPH-dependent cytochrome P450 reductase in the course of electron transfer to cytochromes P450.Biochim Biophys Acta. 2011 Jan;1814(1):132-8. doi: 10.1016/j.bbapap.2010.07.003. Epub 2010 Jul 17. Biochim Biophys Acta. 2011. PMID: 20624491 Review.
-
Cytochrome P450 (CYP) enzymes and the development of CYP biosensors.Biosens Bioelectron. 2013 Jan 15;39(1):1-13. doi: 10.1016/j.bios.2012.05.043. Epub 2012 Jun 26. Biosens Bioelectron. 2013. PMID: 22809523 Review.
Cited by
-
Multiresolution molecular dynamics simulations reveal the interplay between conformational variability and functional interactions in membrane-bound cytochrome P450 2B4.Protein Sci. 2024 Oct;33(10):e5165. doi: 10.1002/pro.5165. Protein Sci. 2024. PMID: 39291728 Free PMC article.
-
Cytochrome P450 Surface Domains Prevent the β-Carotene Monohydroxylase CYP97H1 of Euglena gracilis from Acting as a Dihydroxylase.Biomolecules. 2023 Feb 15;13(2):366. doi: 10.3390/biom13020366. Biomolecules. 2023. PMID: 36830734 Free PMC article.
-
Amphipol-facilitated elucidation of the functional tetrameric complex of full-length cytochrome P450 CYP2B4 and NADPH-cytochrome P450 oxidoreductase.J Biol Chem. 2021 Jan-Jun;296:100645. doi: 10.1016/j.jbc.2021.100645. Epub 2021 Apr 9. J Biol Chem. 2021. PMID: 33839156 Free PMC article.
-
Unraveling the Role of P450 Reductase in Herbicide Metabolic Resistance Mechanism.Plant Direct. 2025 Aug 14;9(8):e70102. doi: 10.1002/pld3.70102. eCollection 2025 Aug. Plant Direct. 2025. PMID: 40823267 Free PMC article.
-
Phylogenetic Analysis and Protein Modelling of Isoflavonoid Synthase Highlights Key Catalytic Sites towards Realising New Bioengineering Endeavours.Bioengineering (Basel). 2022 Oct 24;9(11):609. doi: 10.3390/bioengineering9110609. Bioengineering (Basel). 2022. PMID: 36354520 Free PMC article.
References
-
- Ortiz de Montellano, P. R. in Cytochrome P450: Structure, Mechanism, and Biochemistry (ed. Ortiz de Montellano, P. R.) 111–176 (Springer International Publishing, 2015).
-
- Mustafa, G., Yu, X. & Wade, R. C. in Drug Metabolism Prediction Vol. 38 (ed. Kirchmair, J.) 75–102 (Wiley‐VCH Verlag GmbH & Co. KGaA., 2014).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
