

Significantly improving the quality of genome assemblies through curation
- PMID: 33420778
- PMCID: PMC7794651
- DOI: 10.1093/gigascience/giaa153
Significantly improving the quality of genome assemblies through curation
Abstract
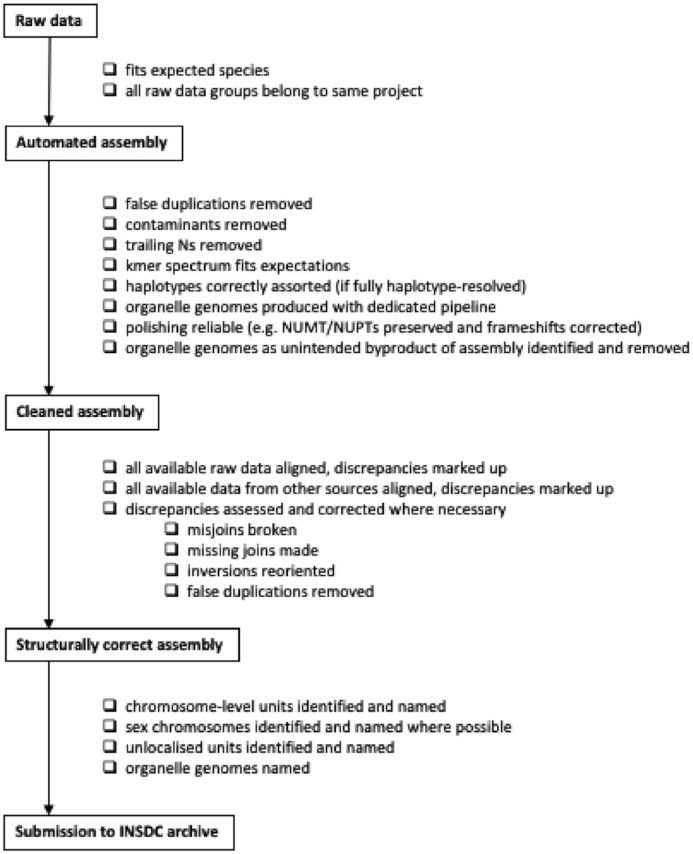
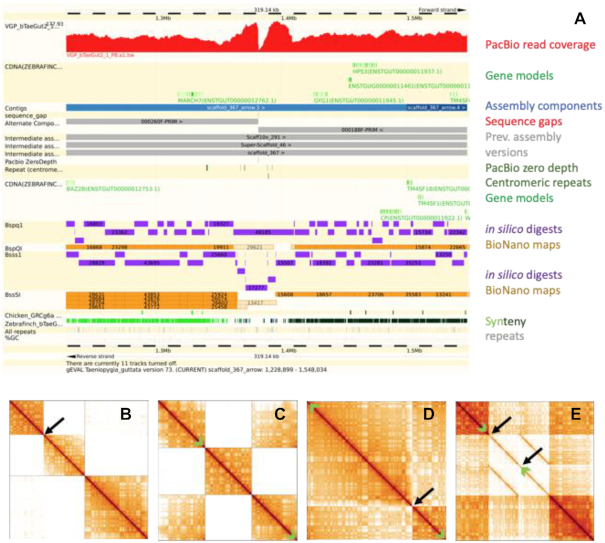
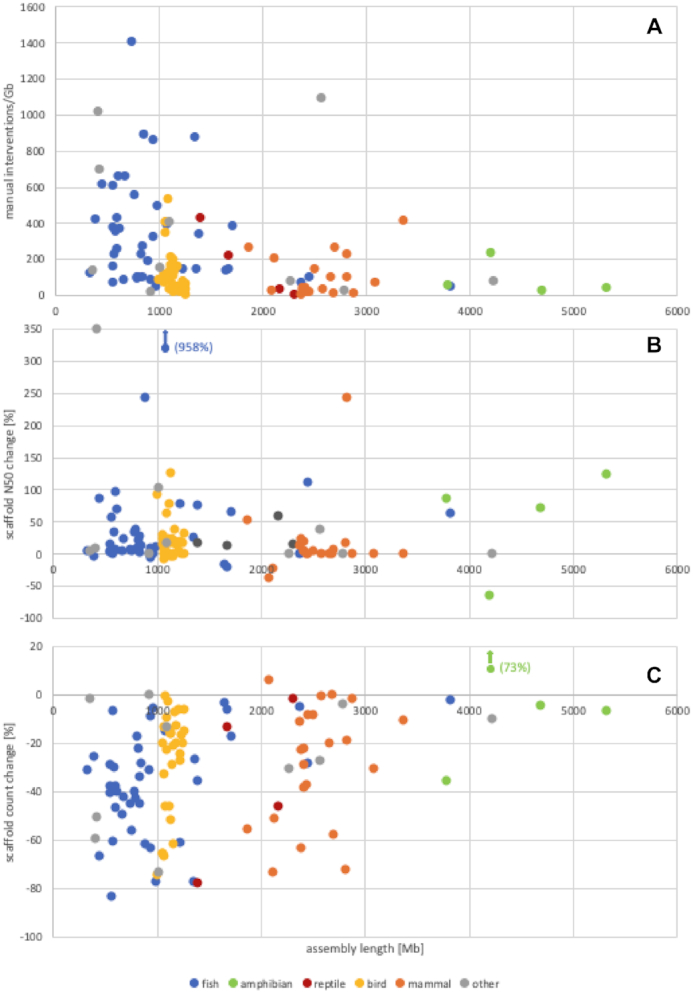
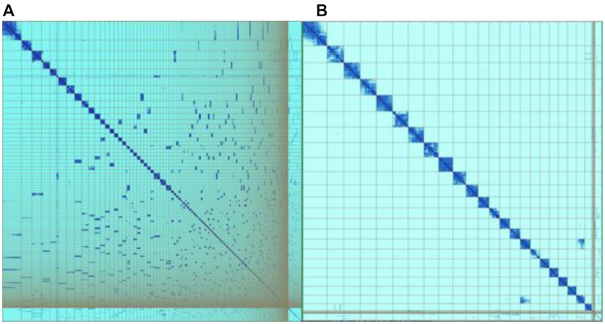
Genome sequence assemblies provide the basis for our understanding of biology. Generating error-free assemblies is therefore the ultimate, but sadly still unachieved goal of a multitude of research projects. Despite the ever-advancing improvements in data generation, assembly algorithms and pipelines, no automated approach has so far reliably generated near error-free genome assemblies for eukaryotes. Whilst working towards improved datasets and fully automated pipelines, assembly evaluation and curation is actively used to bridge this shortcoming and significantly reduce the number of assembly errors. In addition to this increase in product value, the insights gained from assembly curation are fed back into the automated assembly strategy and contribute to notable improvements in genome assembly quality. We describe our tried and tested approach for assembly curation using gEVAL, the genome evaluation browser. We outline the procedures applied to genome curation using gEVAL and also our recommendations for assembly curation in a gEVAL-independent context to facilitate the uptake of genome curation in the wider community.
Keywords: assembly; curation; gEVAL; genome.
© The Author(s) 2021. Published by Oxford University Press GigaScience.
Figures

References
-
- Seppey M, Manni M, Zdobnov EM. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol Biol. 2019;1962:227–45. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
