

Hierarchical Graph Representation of Pharmacophore Models
- PMID: 33425991
- PMCID: PMC7793842
- DOI: 10.3389/fmolb.2020.599059
Hierarchical Graph Representation of Pharmacophore Models
Abstract
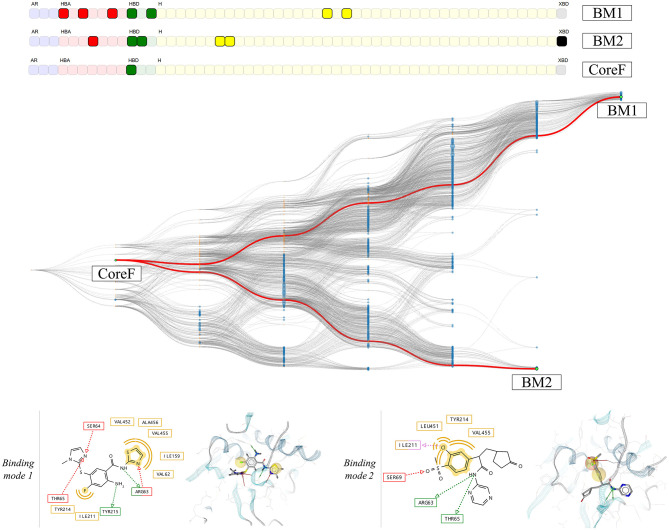
For the investigation of protein-ligand interaction patterns, the current accessibility of a wide variety of sampling methods allows quick access to large-scale data. The main example is the intensive use of molecular dynamics simulations applied to crystallographic structures which provide dynamic information on the binding interactions in protein-ligand complexes. Chemical feature interaction based pharmacophore models extracted from these simulations, were recently used with consensus scoring approaches to identify potentially active molecules. While this approach is rapid and can be fully automated for virtual screening, additional relevant information from such simulations is still opaque and so far the full potential has not been entirely exploited. To address these aspects, we developed the hierarchical graph representation of pharmacophore models (HGPM). This single graph representation enables an intuitive observation of numerous pharmacophore models from long MD trajectories and further emphasizes their relationship and feature hierarchy. The resulting interactive depiction provides an easy-to-apprehend tool for the selection of sets of pharmacophores as well as visual support for analysis of pharmacophore feature composition and virtual screening results. Furthermore, the representation can be adapted to include information involving interactions between the same protein and multiple different ligands. Herein, we describe the generation, visualization and use of HGPMs generated from MD simulations of two x-ray crystallographic derived structures of the human glucokinase protein in complex with allosteric activators. The results demonstrate that a large number of pharmacophores and their relationships can be visualized in an interactive, efficient manner, unique binding modes identified and a combination of models derived from long MD simulations can be strategically prioritized for VS campaigns.
Keywords: clustering; hierarchical graph representation; human glucokinase; molecular dynamic (MD) simulation; pharmacophore modeling; protein ligand binding; protein structure; virtual screening.
Copyright © 2020 Arthur, Oliver, Klaus, Thomas, Gökhan, Sharon, Isabelle, Pierre and Thierry.
Conflict of interest statement
BS and IG are employees of the company Inte:Ligand Software-Entwicklungs und Consulting GmbH, Vienna, Austria. TI and DP are employees of Institut de Recherches Servier (IdRS), Croissy-sur-Seine, France. The herein presented work is the result of a joint research project funded by the aforementioned companies. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures









References
-
- Berthold M. R., Cebron N., Dill F., Fatta G. D., Gabriel T. R., Georg F., et al. (2009). Knime: The Konstanz Information Miner. 4.
LinkOut - more resources
Full Text Sources