

Galactosialidosis: preclinical enzyme replacement therapy in a mouse model of the disease, a proof of concept
- PMID: 33426146
- PMCID: PMC7782203
- DOI: 10.1016/j.omtm.2020.11.012
Galactosialidosis: preclinical enzyme replacement therapy in a mouse model of the disease, a proof of concept
Abstract
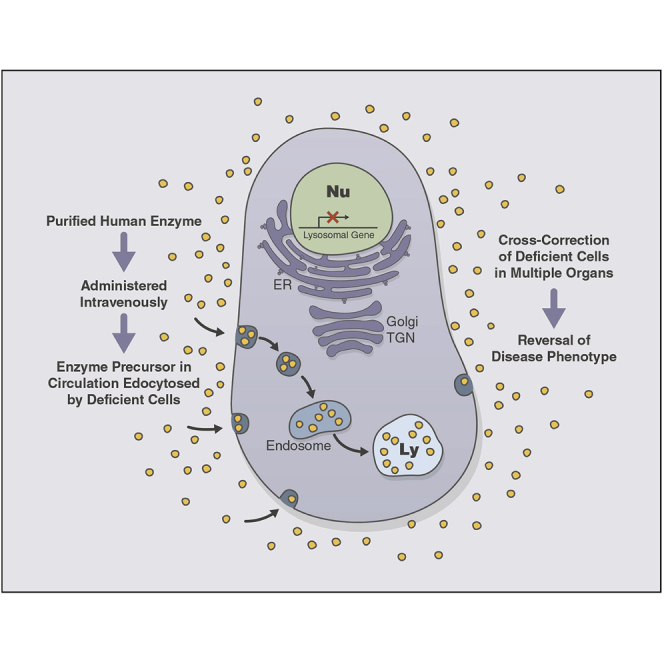
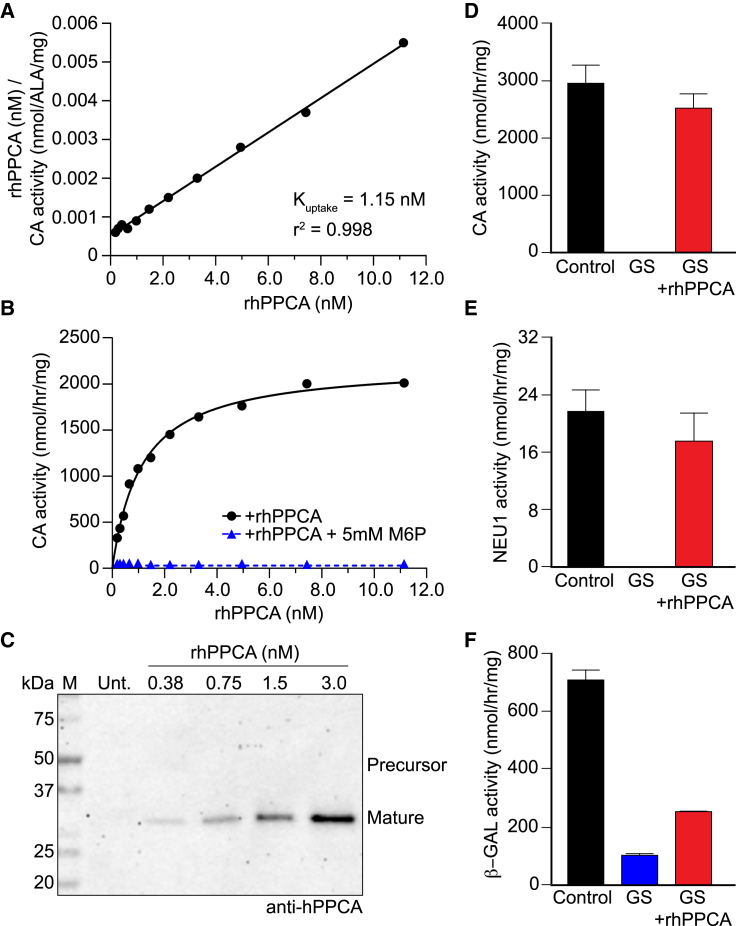
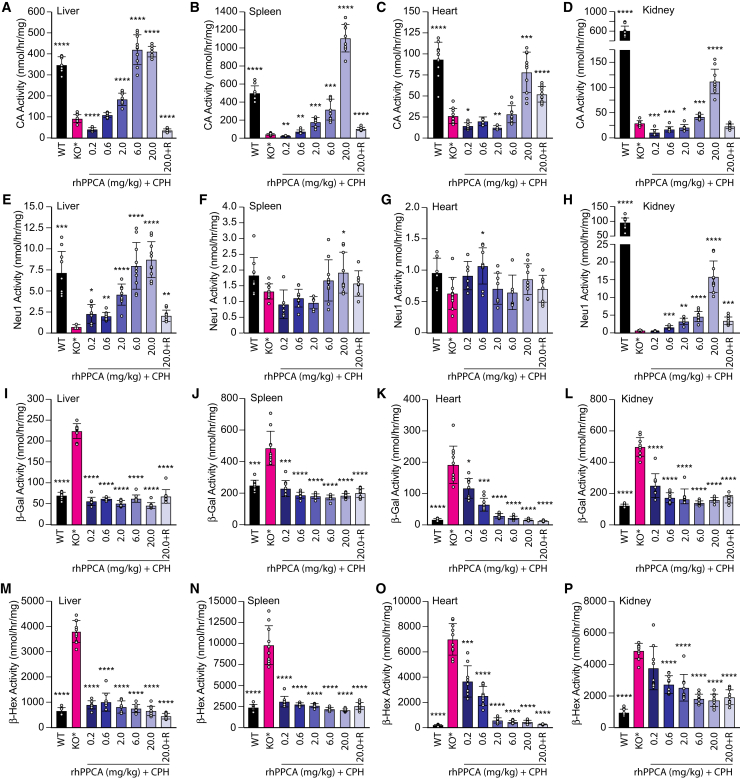
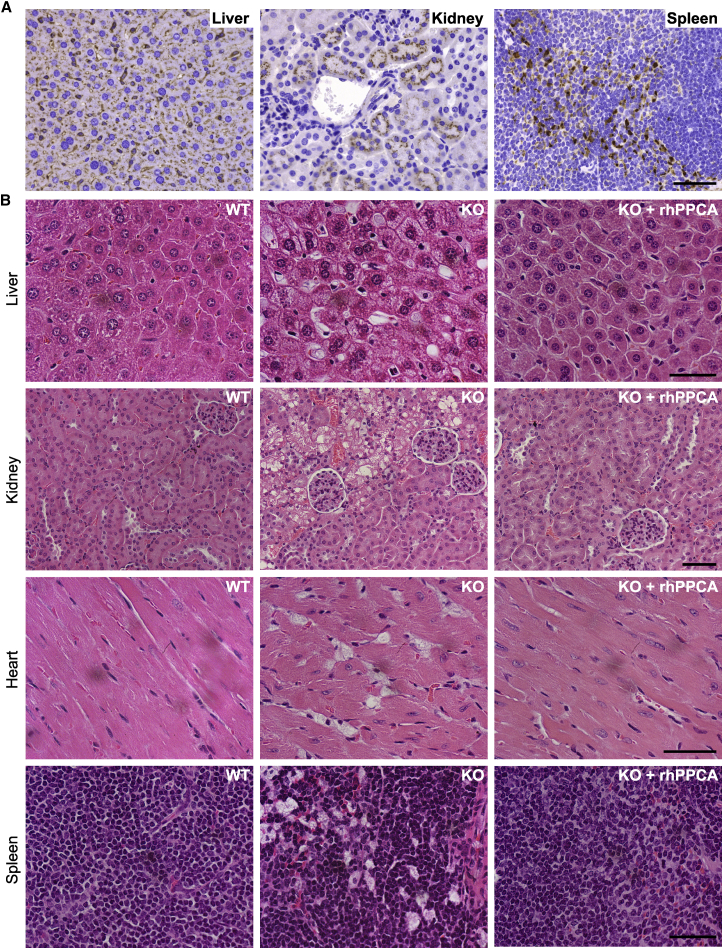
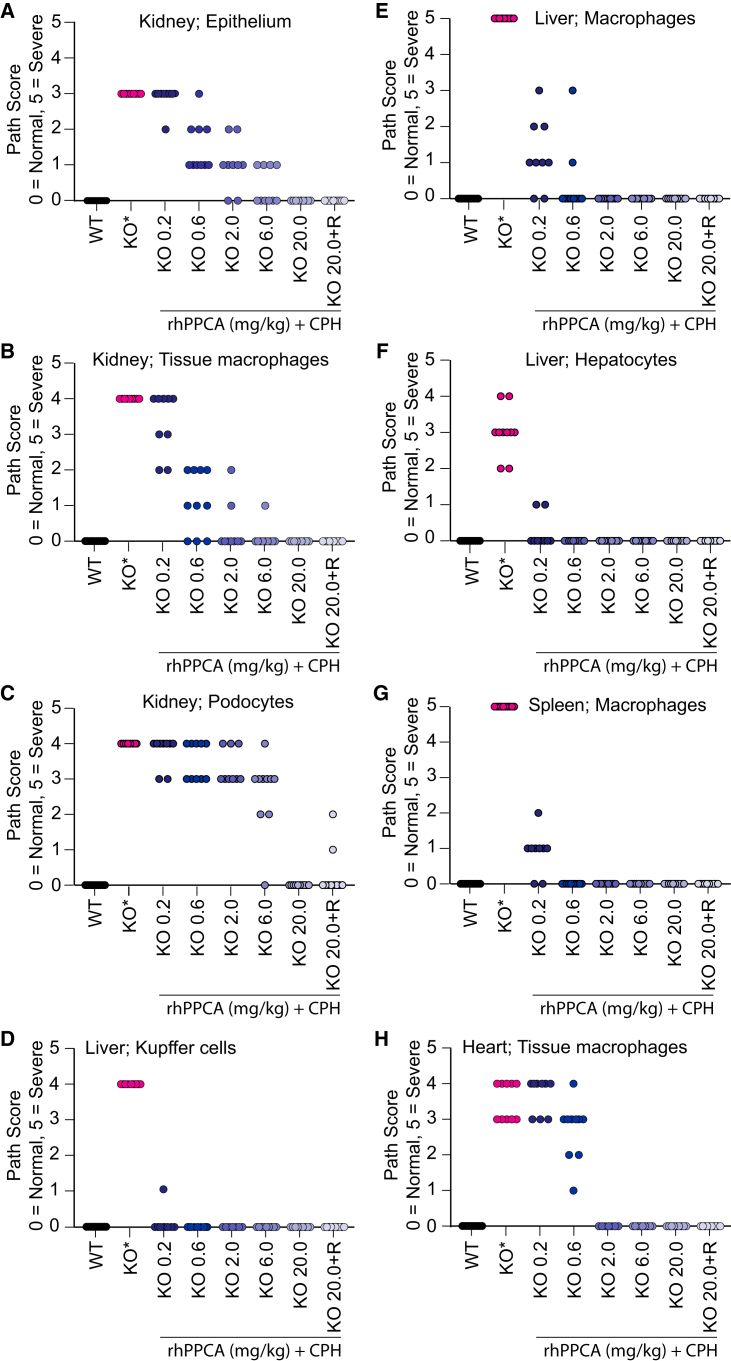
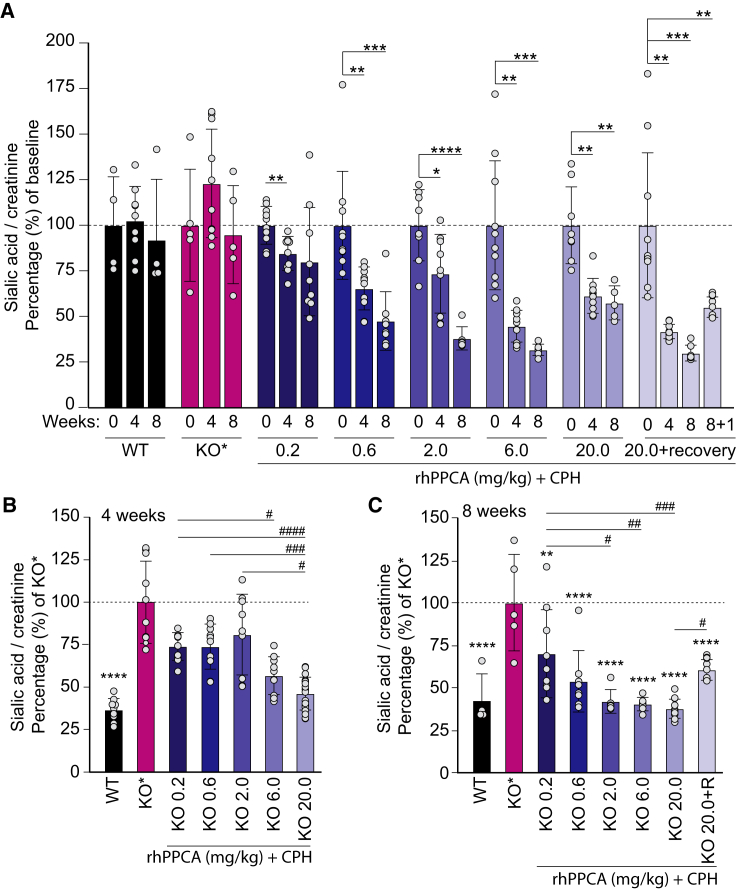
Galactosialidosis is a rare lysosomal storage disease caused by a congenital defect of protective protein/cathepsin A (PPCA) and secondary deficiency of neuraminidase-1 and β-galactosidase. PPCA is a lysosomal serine carboxypeptidase that functions as a chaperone for neuraminidase-1 and β-galactosidase within a lysosomal multi-protein complex. Combined deficiency of the three enzymes leads to accumulation of sialylated glycoproteins and oligosaccharides in tissues and body fluids and manifests in a systemic disease pathology with severity mostly correlating with the type of mutation(s) and age of onset of the symptoms. Here, we describe a proof-of-concept, preclinical study toward the development of enzyme replacement therapy for galactosialidosis, using a recombinant human PPCA. We show that the recombinant enzyme, taken up by patient-derived fibroblasts, restored cathepsin A, neuraminidase-1, and β-galactosidase activities. Long-term, bi-weekly injection of the recombinant enzyme in a cohort of mice with null mutation at the PPCA (CTSA) locus (PPCA -/- ), a faithful model of the disease, demonstrated a dose-dependent, systemic internalization of the enzyme by cells of various organs, including the brain. This resulted in restoration/normalization of the three enzyme activities, resolution of histopathology, and reduction of sialyloligosacchariduria. These positive results underscore the benefits of a PPCA-mediated enzyme replacement therapy for the treatment of galactosialidosis.
Keywords: ERT; galactosialidosis; lysosomal NEU1; lysosomal glycoproteinosis; lysosomal storage disease; lysosomes; recombinant Protective protein/cathepsin A; β-GAL.
© 2020 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Lysosomal sialidase NEU1, its intracellular properties, deficiency, and use as a therapeutic agent.Glycoconj J. 2023 Dec;40(6):611-619. doi: 10.1007/s10719-023-10135-6. Epub 2023 Dec 26. Glycoconj J. 2023. PMID: 38147151 Review.
-
Intermittent enzyme replacement therapy with recombinant human β-galactosidase prevents neuraminidase 1 deficiency.J Biol Chem. 2020 Sep 25;295(39):13556-13569. doi: 10.1074/jbc.RA119.010794. Epub 2020 Jul 28. J Biol Chem. 2020. PMID: 32727849 Free PMC article.
-
Galactosialidosis: historic aspects and overview of investigated and emerging treatment options.Expert Opin Orphan Drugs. 2017;5(2):131-141. doi: 10.1080/21678707.2016.1266933. Epub 2016 Dec 14. Expert Opin Orphan Drugs. 2017. PMID: 28603679 Free PMC article.
-
Targeting macrophages with baculovirus-produced lysosomal enzymes: implications for enzyme replacement therapy of the glycoprotein storage disorder galactosialidosis.FASEB J. 2004 Jun;18(9):971-3. doi: 10.1096/fj.03-0941fje. Epub 2004 Apr 14. FASEB J. 2004. PMID: 15084520
-
Cathepsin A/protective protein: an unusual lysosomal multifunctional protein.Cell Mol Life Sci. 1999 Dec;56(11-12):894-907. doi: 10.1007/s000180050482. Cell Mol Life Sci. 1999. PMID: 11212324 Free PMC article. Review.
Cited by
-
Preclinical Enzyme Replacement Therapy with a Recombinant β-Galactosidase-Lectin Fusion for CNS Delivery and Treatment of GM1-Gangliosidosis.Cells. 2022 Aug 19;11(16):2579. doi: 10.3390/cells11162579. Cells. 2022. PMID: 36010656 Free PMC article.
-
Galactosialidosis: A Report of Three Cases Diagnosed With a Founder Genetic Mutation in the Bahraini Population.Cureus. 2025 Jan 20;17(1):e77750. doi: 10.7759/cureus.77750. eCollection 2025 Jan. Cureus. 2025. PMID: 39981487 Free PMC article.
-
Inflammatory arthritis complicating galactosialidosis: a case report.BMC Rheumatol. 2021 Oct 11;5(1):41. doi: 10.1186/s41927-021-00208-0. BMC Rheumatol. 2021. PMID: 34629108 Free PMC article.
-
Structure of the immunoregulatory sialidase NEU1.Sci Adv. 2023 May 19;9(20):eadf8169. doi: 10.1126/sciadv.adf8169. Epub 2023 May 19. Sci Adv. 2023. PMID: 37205763 Free PMC article.
-
Lysosomal sialidase NEU1, its intracellular properties, deficiency, and use as a therapeutic agent.Glycoconj J. 2023 Dec;40(6):611-619. doi: 10.1007/s10719-023-10135-6. Epub 2023 Dec 26. Glycoconj J. 2023. PMID: 38147151 Review.
References
-
- d’Azzo A., Andria G., Bonten E., Annunziata I. Volume 183. McGraw-Hill Medical; 2014. Galactosialidosis. The Online Metabolic and Molecular Bases of Inherited Disease.
-
- Galjart N.J., Gillemans N., Harris A., van der Horst G.T., Verheijen F.W., Galjaard H., d’Azzo A. Expression of cDNA encoding the human “protective protein” associated with lysosomal beta-galactosidase and neuraminidase: homology to yeast proteases. Cell. 1988;54:755–764. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
