

Current Concepts and Advances in Graft-Versus-Host Disease Immunology
- PMID: 33428454
- PMCID: PMC8085043
- DOI: 10.1146/annurev-immunol-102119-073227
Current Concepts and Advances in Graft-Versus-Host Disease Immunology
Abstract
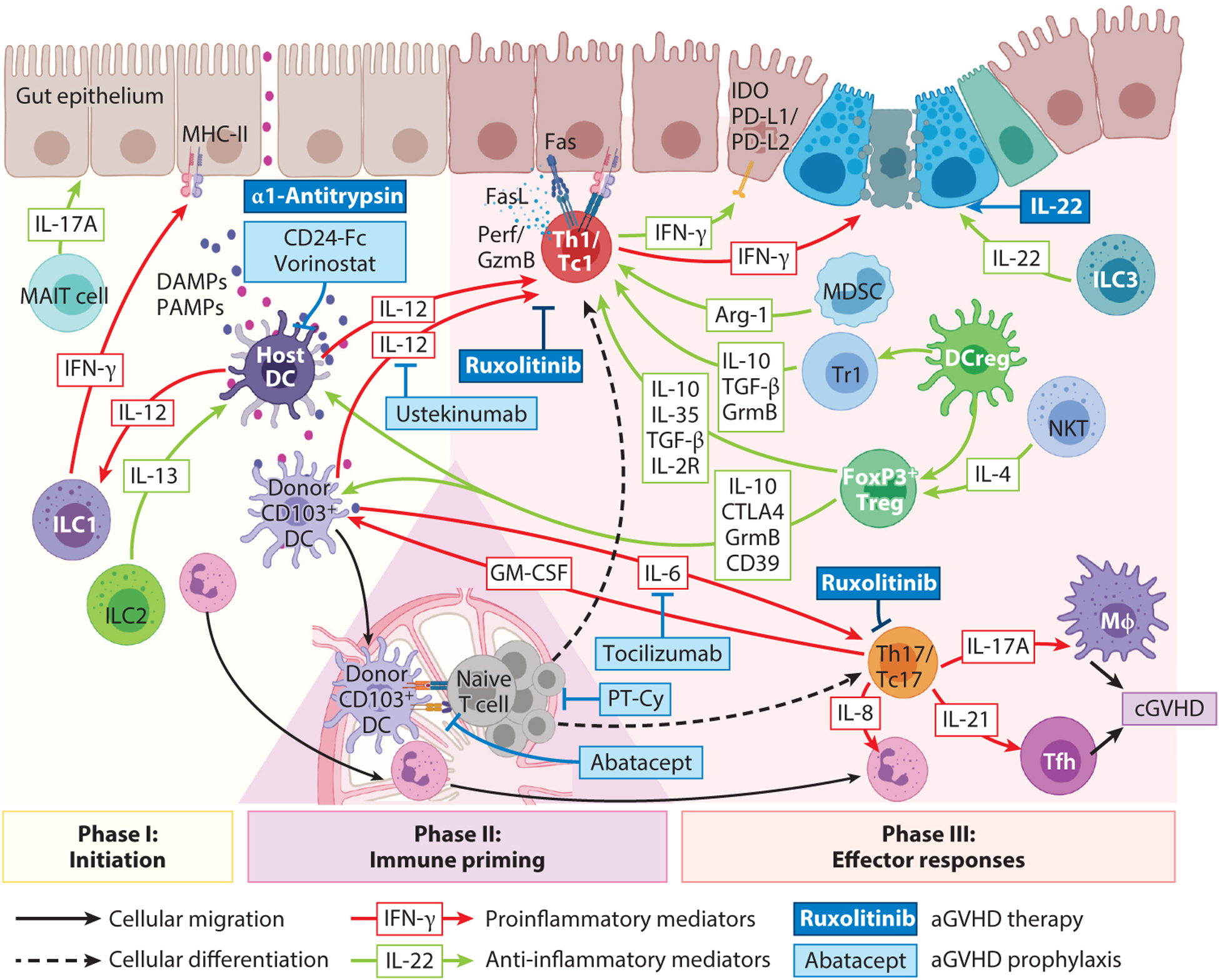
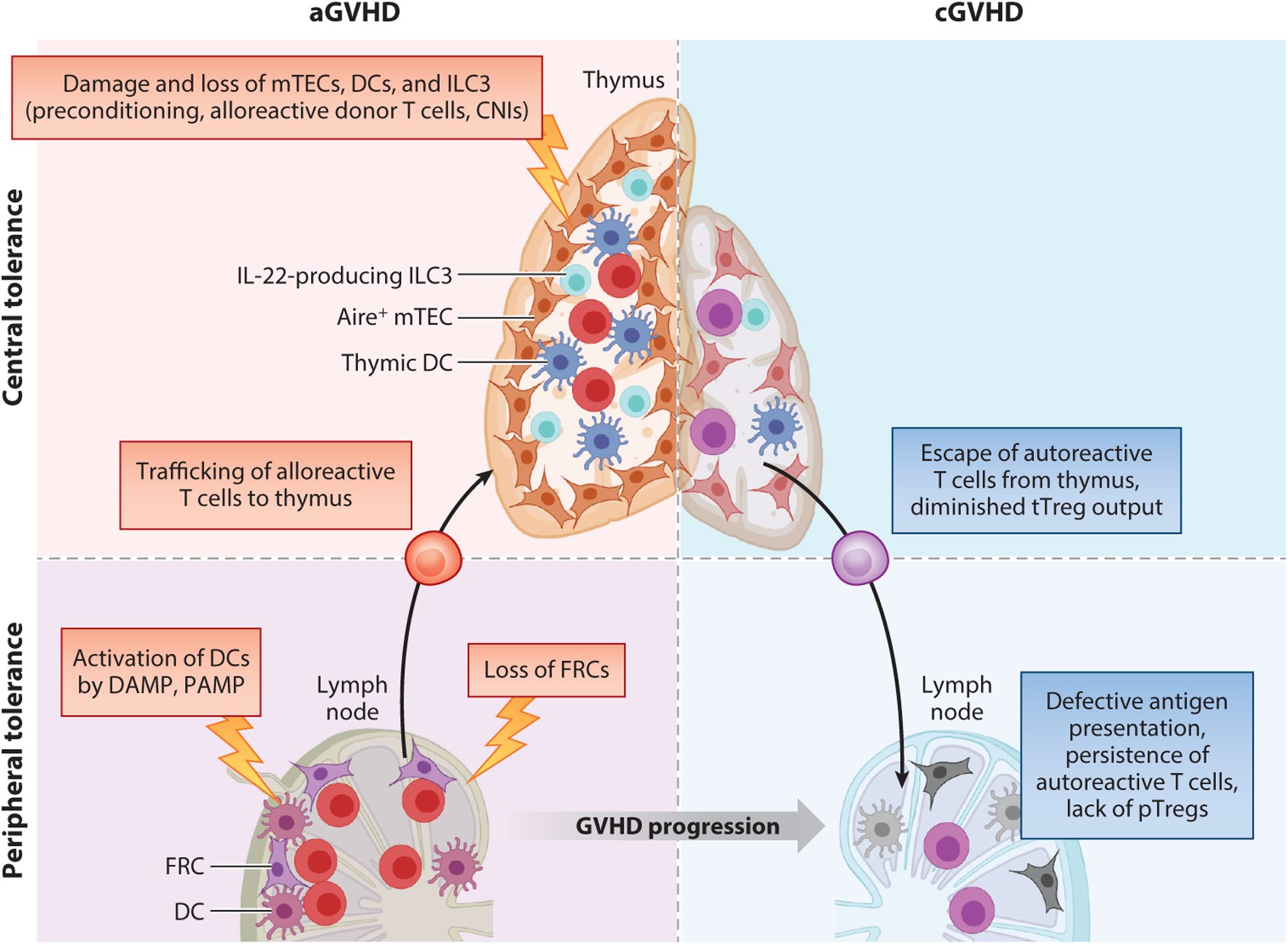
Worldwide, each year over 30,000 patients undergo an allogeneic hema-topoietic stem cell transplantation with the intent to cure high-risk hematologic malignancy, immunodeficiency, metabolic disease, or a life-threatening bone marrow failure syndrome. Despite substantial advances in donor selection and conditioning regimens and greater availability of allograft sources, transplant recipients still endure the morbidity and mortality of graft-versus-host disease (GVHD). Herein, we identify key aspects of acute and chronic GVHD pathophysiology, including host/donor cell effectors, gut dysbiosis, immune system and cytokine imbalance, and the interface between inflammation and tissue fibrosis. In particular, we also summarize the translational application of this heightened understanding of immune dysregulation in the design of novel therapies to prevent and treat GVHD.
Keywords: graft-versus-host disease; pathophysiology; prevention; regulation; therapy; tolerance.
Figures

References
-
- Zeiser R, Blazar BR. 2017. Pathophysiology of chronic graft-versus-host disease and therapeutic targets. N. Engl. J. Med 377:2565–79 - PubMed
Publication types
MeSH terms
Grants and funding
- R01 HL133823/HL/NHLBI NIH HHS/United States
- R01 HL095791/HL/NHLBI NIH HHS/United States
- R01 FD004099/FD/FDA HHS/United States
- P01 CA065493/CA/NCI NIH HHS/United States
- R01 HL118979/HL/NHLBI NIH HHS/United States
- U19 AI051731/AI/NIAID NIH HHS/United States
- R01 HL147324/HL/NHLBI NIH HHS/United States
- R37 AI034495/AI/NIAID NIH HHS/United States
- R01 HL155114/HL/NHLBI NIH HHS/United States
- R01 HL056067/HL/NHLBI NIH HHS/United States
- U01 CA244291/CA/NCI NIH HHS/United States
- P01 AI056299/AI/NIAID NIH HHS/United States
- R01 HL148164/HL/NHLBI NIH HHS/United States
- P01 CA142106/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
