

CReM: chemically reasonable mutations framework for structure generation
- PMID: 33430959
- PMCID: PMC7178718
- DOI: 10.1186/s13321-020-00431-w
CReM: chemically reasonable mutations framework for structure generation
Abstract
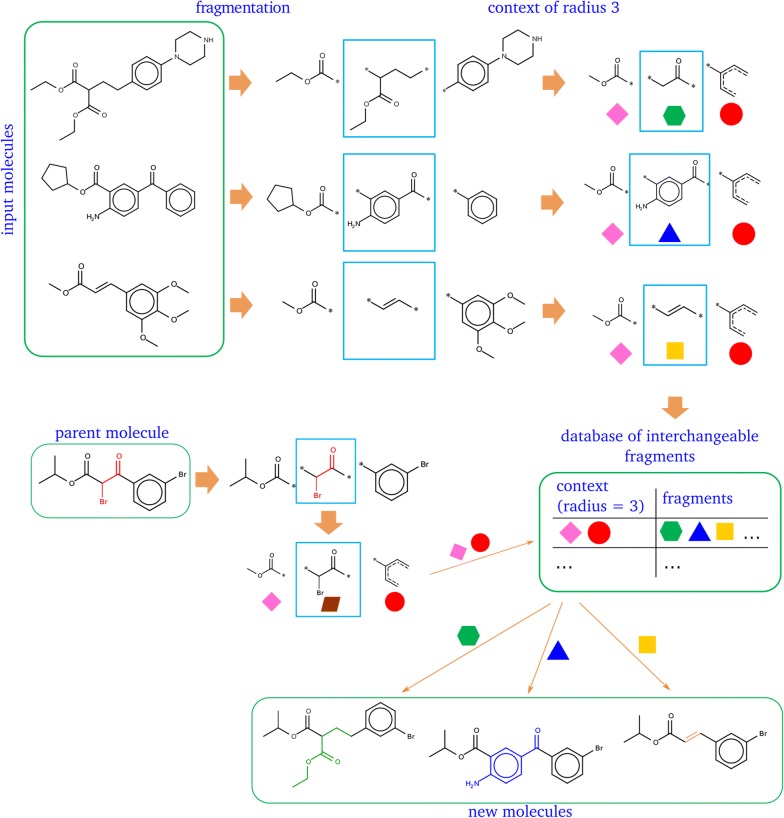
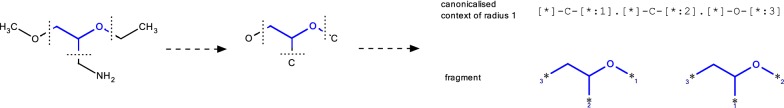
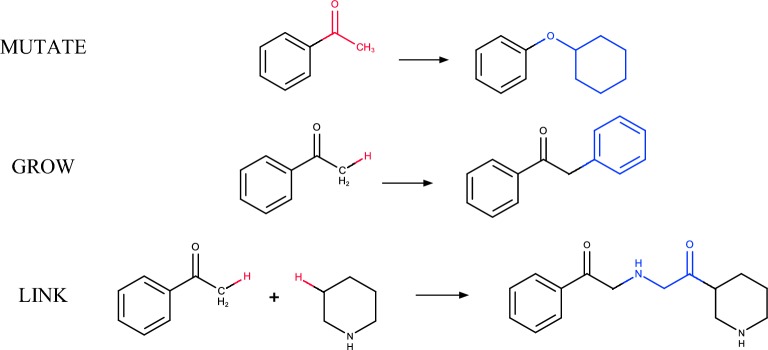
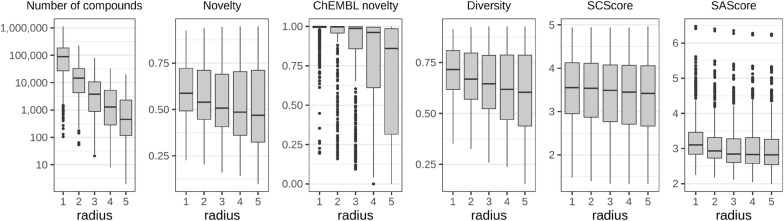
Structure generators are widely used in de novo design studies and their performance substantially influences an outcome. Approaches based on the deep learning models and conventional atom-based approaches may result in invalid structures and fail to address their synthetic feasibility issues. On the other hand, conventional reaction-based approaches result in synthetically feasible compounds but novelty and diversity of generated compounds may be limited. Fragment-based approaches can provide both better novelty and diversity of generated compounds but the issue of synthetic complexity of generated structure was not explicitly addressed before. Here we developed a new framework of fragment-based structure generation that, by design, results in the chemically valid structures and provides flexible control over diversity, novelty, synthetic complexity and chemotypes of generated compounds. The framework was implemented as an open-source Python module and can be used to create custom workflows for the exploration of chemical space.
Keywords: De novo design; De novo structure generation; Matched molecular pairs.
Conflict of interest statement
The author declares no competing interests.
Figures
Similar articles
-
Control of Synthetic Feasibility of Compounds Generated with CReM.J Chem Inf Model. 2020 Dec 28;60(12):6074-6080. doi: 10.1021/acs.jcim.0c00792. Epub 2020 Nov 9. J Chem Inf Model. 2020. PMID: 33167612
-
LOGICS: Learning optimal generative distribution for designing de novo chemical structures.J Cheminform. 2023 Sep 7;15(1):77. doi: 10.1186/s13321-023-00747-3. J Cheminform. 2023. PMID: 37674239 Free PMC article.
-
NAOMInext - Synthetically feasible fragment growing in a structure-based design context.Eur J Med Chem. 2019 Feb 1;163:747-762. doi: 10.1016/j.ejmech.2018.11.075. Epub 2018 Dec 6. Eur J Med Chem. 2019. PMID: 30576905
-
Scaffold-hopping potential of fragment-based de novo design: the chances and limits of variation.Comb Chem High Throughput Screen. 2009 May;12(4):383-96. doi: 10.2174/138620709788167971. Comb Chem High Throughput Screen. 2009. PMID: 19442066 Review.
-
De novo design: balancing novelty and confined chemical space.Expert Opin Drug Discov. 2010 Aug;5(8):789-812. doi: 10.1517/17460441.2010.497534. Epub 2010 Jun 30. Expert Opin Drug Discov. 2010. PMID: 22827800 Review.
Cited by
-
Parallel tempered genetic algorithm guided by deep neural networks for inverse molecular design.Digit Discov. 2022 May 3;1(4):390-404. doi: 10.1039/d2dd00003b. eCollection 2022 Aug 8. Digit Discov. 2022. PMID: 36091415 Free PMC article.
-
MolPLA: a molecular pretraining framework for learning cores, R-groups and their linker joints.Bioinformatics. 2024 Jun 28;40(Suppl 1):i369-i380. doi: 10.1093/bioinformatics/btae256. Bioinformatics. 2024. PMID: 38940143 Free PMC article.
-
A molecule perturbation software library and its application to study the effects of molecular design constraints.J Cheminform. 2023 Sep 26;15(1):89. doi: 10.1186/s13321-023-00761-5. J Cheminform. 2023. PMID: 37752561 Free PMC article.
-
Generative Models as an Emerging Paradigm in the Chemical Sciences.J Am Chem Soc. 2023 Apr 26;145(16):8736-8750. doi: 10.1021/jacs.2c13467. Epub 2023 Apr 13. J Am Chem Soc. 2023. PMID: 37052978 Free PMC article. Review.
-
Molecular fragmentation as a crucial step in the AI-based drug development pathway.Commun Chem. 2024 Feb 1;7(1):20. doi: 10.1038/s42004-024-01109-2. Commun Chem. 2024. PMID: 38302655 Free PMC article. Review.
References
-
- Wang R, Gao Y, Lai L. LigBuilder: a multi-purpose program for structure-based drug design. Mol Model Annu. 2000;6:498–516. doi: 10.1007/s0089400060498. - DOI
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
