

Chemical space exploration based on recurrent neural networks: applications in discovering kinase inhibitors
- PMID: 33430983
- PMCID: PMC7278228
- DOI: 10.1186/s13321-020-00446-3
Chemical space exploration based on recurrent neural networks: applications in discovering kinase inhibitors
Abstract
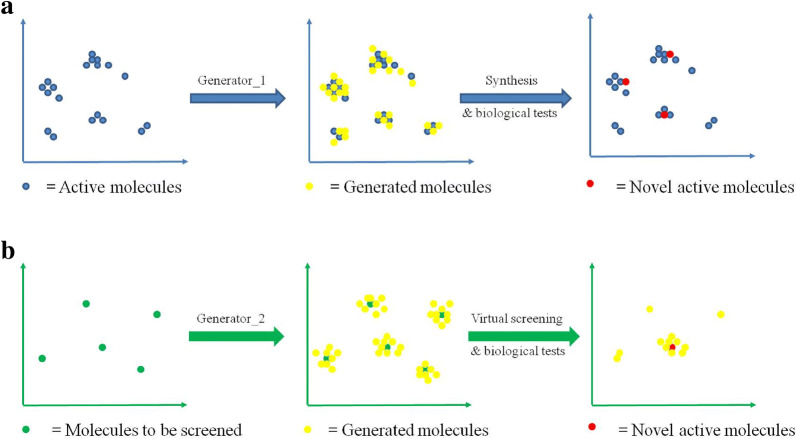
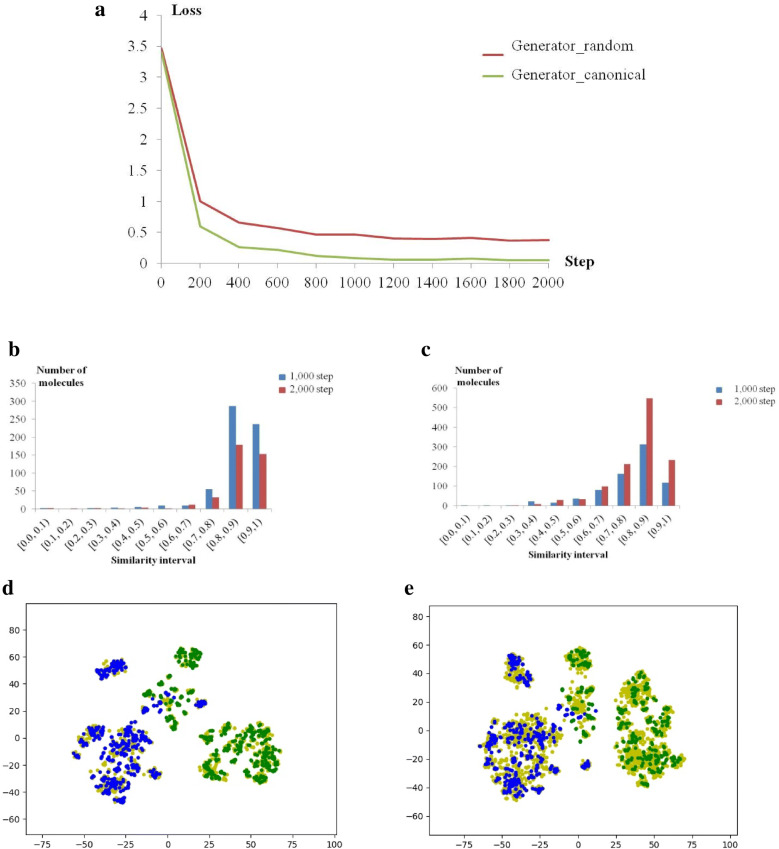
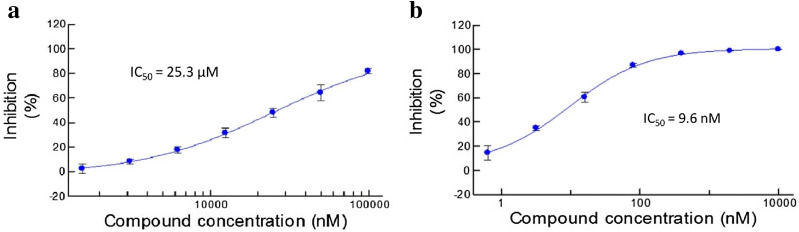
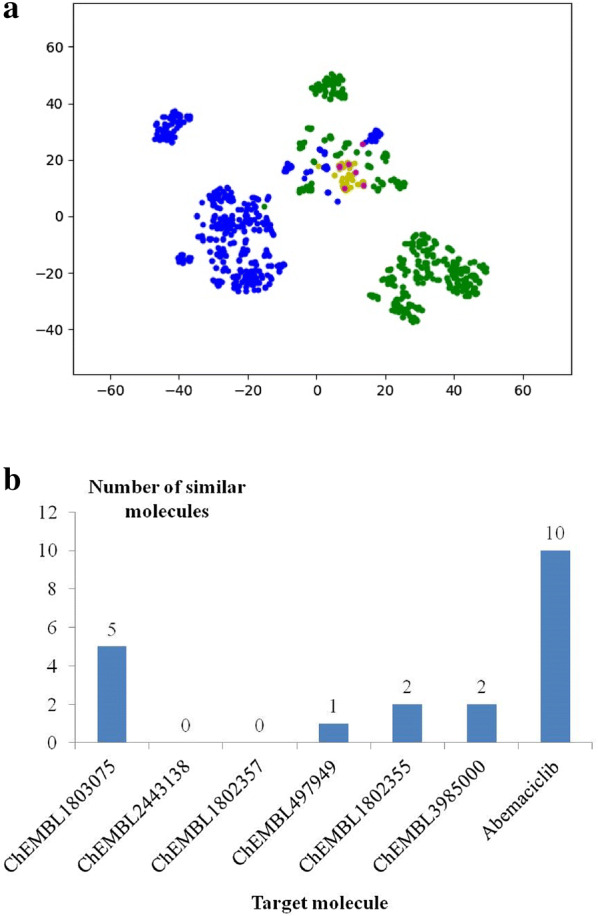
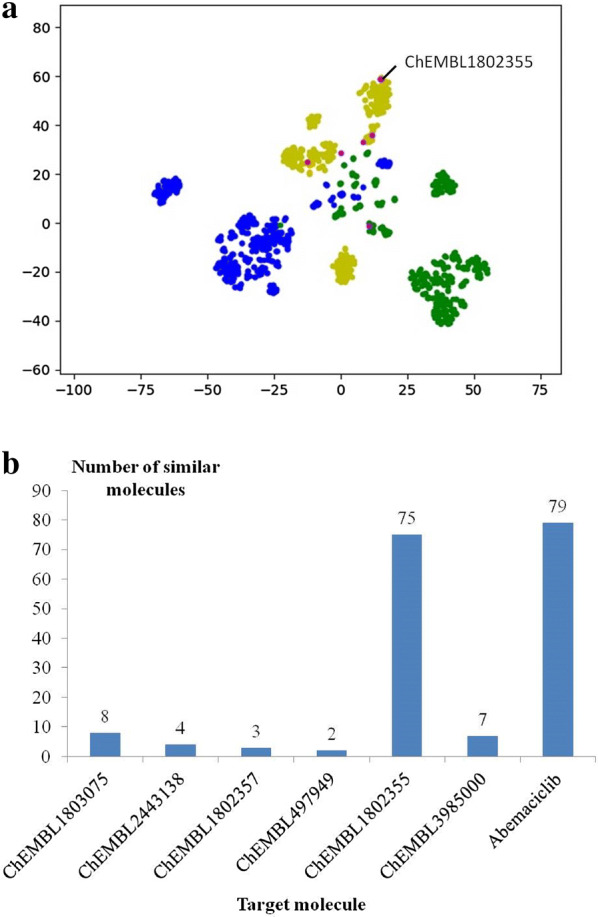
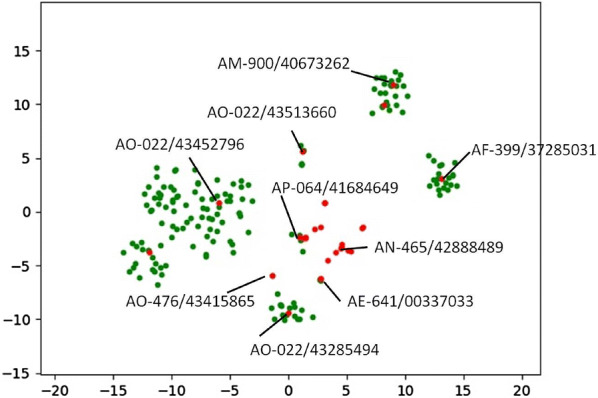
With the rise of artificial intelligence (AI) in drug discovery, de novo molecular generation provides new ways to explore chemical space. However, because de novo molecular generation methods rely on abundant known molecules, generated molecules may have a problem of novelty. Novelty is important in highly competitive areas of medicinal chemistry, such as the discovery of kinase inhibitors. In this study, de novo molecular generation based on recurrent neural networks was applied to discover a new chemical space of kinase inhibitors. During the application, the practicality was evaluated, and new inspiration was found. With the successful discovery of one potent Pim1 inhibitor and two lead compounds that inhibit CDK4, AI-based molecular generation shows potentials in drug discovery and development.
Keywords: Chemical space; De novo molecular generation; Kinase inhibitors; Recurrent neural networks.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Lipinski C, Hopkins A. Navigating chemical space for biology and medicine. Nature. 2004;432(7019):855–861. - PubMed
-
- Mullard A. The drug-maker’s guide to the galaxy. Nature. 2017;549(7673):445–447. - PubMed
-
- Baig MH, Ahmad K, Roy S, Ashraf JM, Adil M, Siddiqui MH, Khan S, Kamal MA, Provaznik I, Choi I. Computer aided drug design: success and limitations. Curr Pharm Des. 2016;22(5):572–581. - PubMed
-
- Schneider G. Automating drug discovery. Nat Rev Drug Discov. 2018;17(2):97–113. - PubMed
-
- Saikin SK, Kreisbeck C, Sheberla D, Becker JS, Aspuru-Guzik A. Closed-loop discovery platform integration is needed for artificial intelligence to make an impact in drug discovery. Expert Opin Drug Discov. 2019;14(1):1–4. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
