

kGCN: a graph-based deep learning framework for chemical structures
- PMID: 33430993
- PMCID: PMC7216578
- DOI: 10.1186/s13321-020-00435-6
kGCN: a graph-based deep learning framework for chemical structures
Abstract
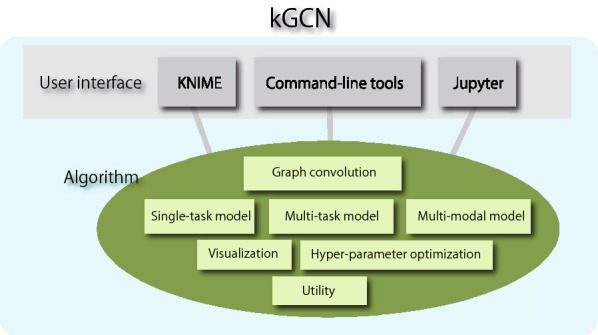
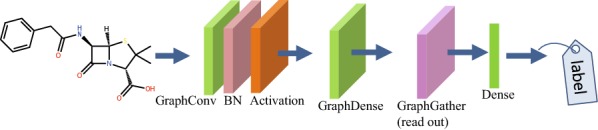
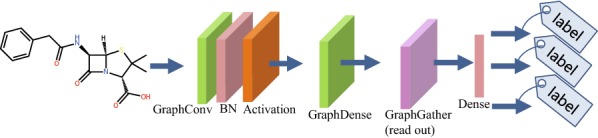
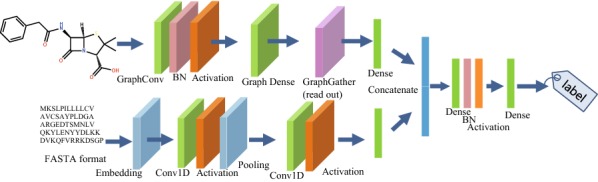
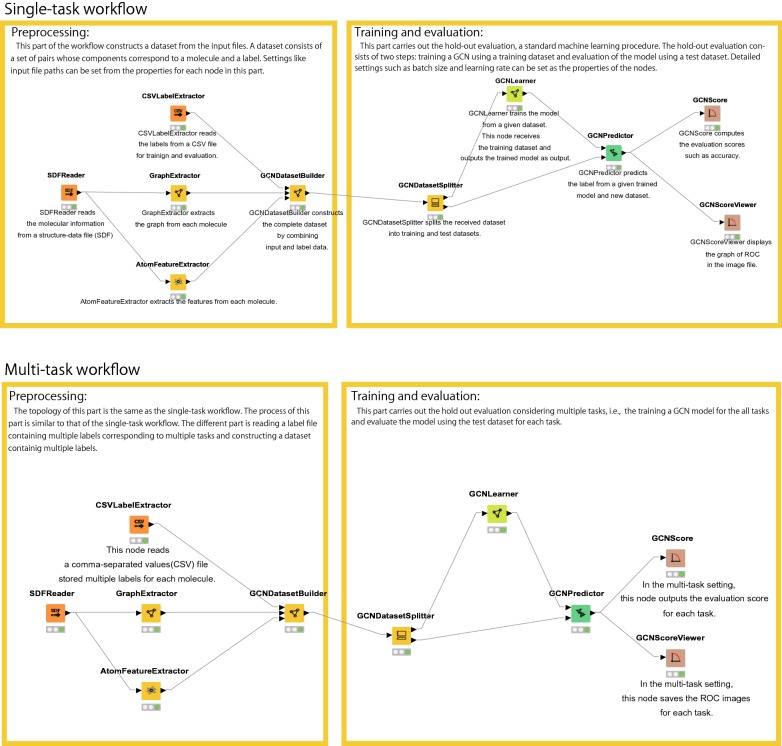
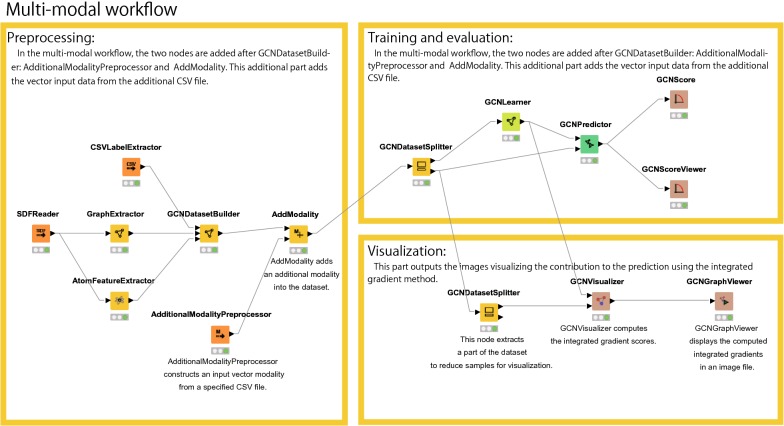
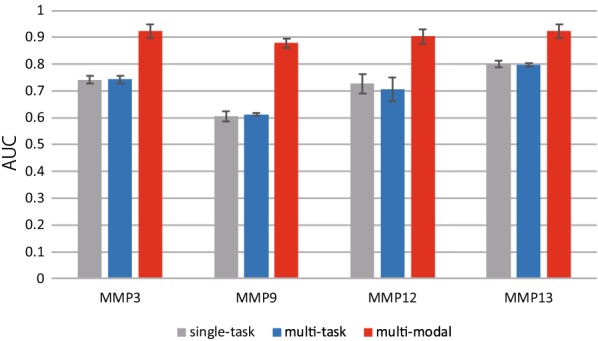
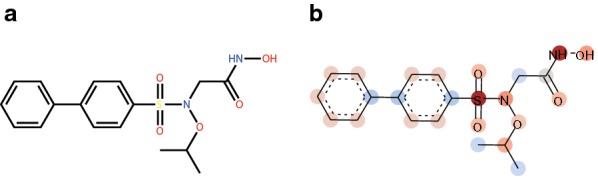
Deep learning is developing as an important technology to perform various tasks in cheminformatics. In particular, graph convolutional neural networks (GCNs) have been reported to perform well in many types of prediction tasks related to molecules. Although GCN exhibits considerable potential in various applications, appropriate utilization of this resource for obtaining reasonable and reliable prediction results requires thorough understanding of GCN and programming. To leverage the power of GCN to benefit various users from chemists to cheminformaticians, an open-source GCN tool, kGCN, is introduced. To support the users with various levels of programming skills, kGCN includes three interfaces: a graphical user interface (GUI) employing KNIME for users with limited programming skills such as chemists, as well as command-line and Python library interfaces for users with advanced programming skills such as cheminformaticians. To support the three steps required for building a prediction model, i.e., pre-processing, model tuning, and interpretation of results, kGCN includes functions of typical pre-processing, Bayesian optimization for automatic model tuning, and visualization of the atomic contribution to prediction for interpretation of results. kGCN supports three types of approaches, single-task, multi-task, and multi-modal predictions. The prediction of compound-protein interaction for four matrixmetalloproteases, MMP-3, -9, -12 and -13, in the inhibition assays is performed as a representative case study using kGCN. Additionally, kGCN provides the visualization of atomic contributions to the prediction. Such visualization is useful for the validation of the prediction models and the design of molecules based on the prediction model, realizing "explainable AI" for understanding the factors affecting AI prediction. kGCN is available at https://github.com/clinfo.
Keywords: Graph convolutional network; Graph neural network; KNIME; Open source software; kGCN.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
A Knowledge-Graph-Based Multimodal Deep Learning Framework for Identifying Drug-Drug Interactions.Molecules. 2023 Feb 3;28(3):1490. doi: 10.3390/molecules28031490. Molecules. 2023. PMID: 36771157 Free PMC article.
-
DGL-LifeSci: An Open-Source Toolkit for Deep Learning on Graphs in Life Science.ACS Omega. 2021 Oct 5;6(41):27233-27238. doi: 10.1021/acsomega.1c04017. eCollection 2021 Oct 19. ACS Omega. 2021. PMID: 34693143 Free PMC article.
-
Pre-training graph neural networks for link prediction in biomedical networks.Bioinformatics. 2022 Apr 12;38(8):2254-2262. doi: 10.1093/bioinformatics/btac100. Bioinformatics. 2022. PMID: 35171981
-
NeuroPycon: An open-source python toolbox for fast multi-modal and reproducible brain connectivity pipelines.Neuroimage. 2020 Oct 1;219:117020. doi: 10.1016/j.neuroimage.2020.117020. Epub 2020 Jun 6. Neuroimage. 2020. PMID: 32522662
-
An inductive graph neural network model for compound-protein interaction prediction based on a homogeneous graph.Brief Bioinform. 2022 May 13;23(3):bbac073. doi: 10.1093/bib/bbac073. Brief Bioinform. 2022. PMID: 35275993 Free PMC article. Review.
Cited by
-
VGAE-MCTS: A New Molecular Generative Model Combining the Variational Graph Auto-Encoder and Monte Carlo Tree Search.J Chem Inf Model. 2023 Dec 11;63(23):7392-7400. doi: 10.1021/acs.jcim.3c01220. Epub 2023 Nov 22. J Chem Inf Model. 2023. PMID: 37993764 Free PMC article.
-
AI-driven prediction of bitterness and sweetness and analysis of receptor interactions.Curr Res Food Sci. 2025 May 19;10:101090. doi: 10.1016/j.crfs.2025.101090. eCollection 2025. Curr Res Food Sci. 2025. PMID: 40497229 Free PMC article.
-
Feature attention graph neural network for estimating brain age and identifying important neural connections in mouse models of genetic risk for Alzheimer's disease.Imaging Neurosci (Camb). 2024 Jul 31;2:imag-2-00245. doi: 10.1162/imag_a_00245. eCollection 2024. Imaging Neurosci (Camb). 2024. PMID: 40800544 Free PMC article.
-
High-Throughput Screening of Promising Redox-Active Molecules with MolGAT.ACS Omega. 2023 Jun 30;8(27):24268-24278. doi: 10.1021/acsomega.3c01295. eCollection 2023 Jul 11. ACS Omega. 2023. PMID: 37457475 Free PMC article.
-
Music recommendation algorithms based on knowledge graph and multi-task feature learning.Sci Rep. 2024 Jan 24;14(1):2055. doi: 10.1038/s41598-024-52463-z. Sci Rep. 2024. PMID: 38267571 Free PMC article.
References
-
- Gawehn E, Hiss JA, Schneider G. Deep learning in drug discovery. Mol Inform. 2016;35(1):3–14. - PubMed
-
- Goh GB, Hodas NO, Vishnu A. Deep learning for computational chemistry. J Comput Chem. 2017;38(16):1291–1307. - PubMed
-
- Elton DC, Boukouvalas Z, Fuge MD, Chung PW. Deep learning for molecular design - a review of the state of the art. Mol Syst Design Eng. 2019;4(4):828–849.
-
- Torng W, Altman RB. Graph convolutional neural networks for predicting drug-target interactions. J Chem Inform Model. 2019;59(10):4131–4149. - PubMed
-
- Ma J, Sheridan RP, Liaw A, Dahl GE, Svetnik V. Deep neural nets as a method for quantitative structure-activity relationships. J Chem Inform Model. 2015;55(2):263–274. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
