

DeepGraphMolGen, a multi-objective, computational strategy for generating molecules with desirable properties: a graph convolution and reinforcement learning approach
- PMID: 33431037
- PMCID: PMC7487898
- DOI: 10.1186/s13321-020-00454-3
DeepGraphMolGen, a multi-objective, computational strategy for generating molecules with desirable properties: a graph convolution and reinforcement learning approach
Abstract
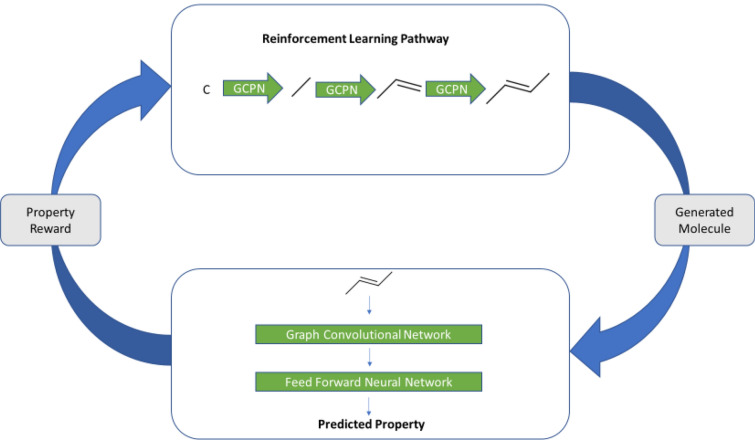
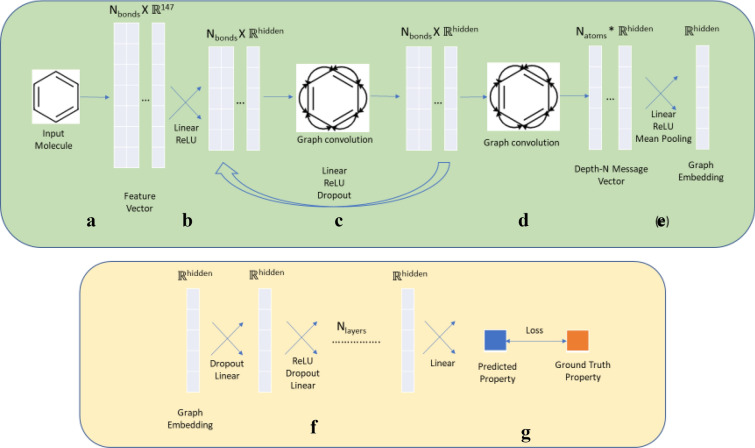
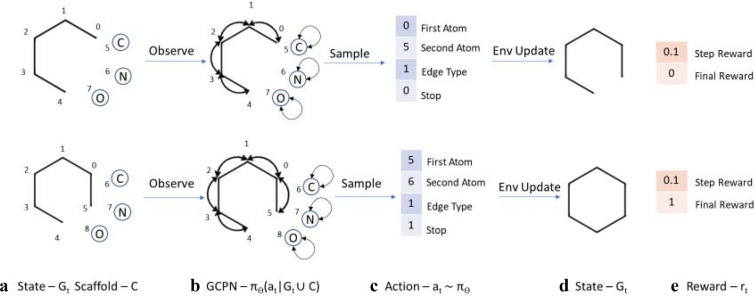
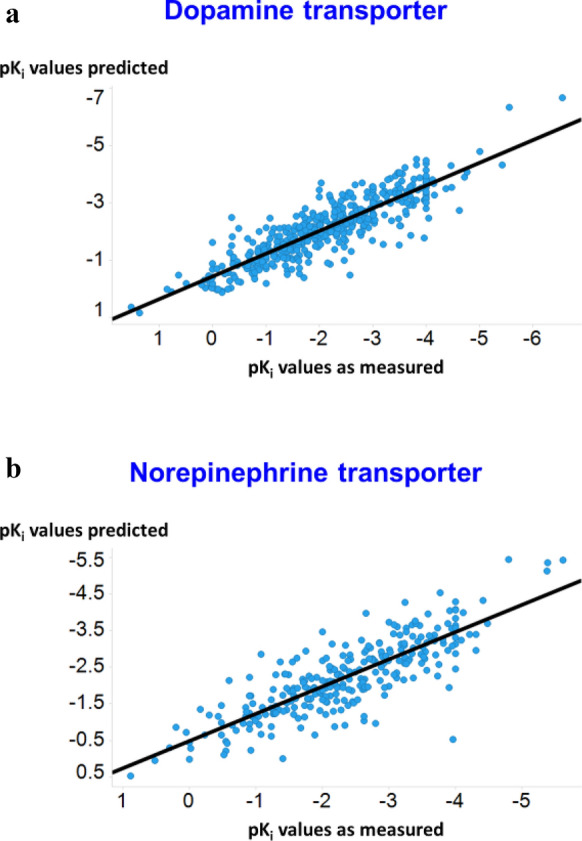
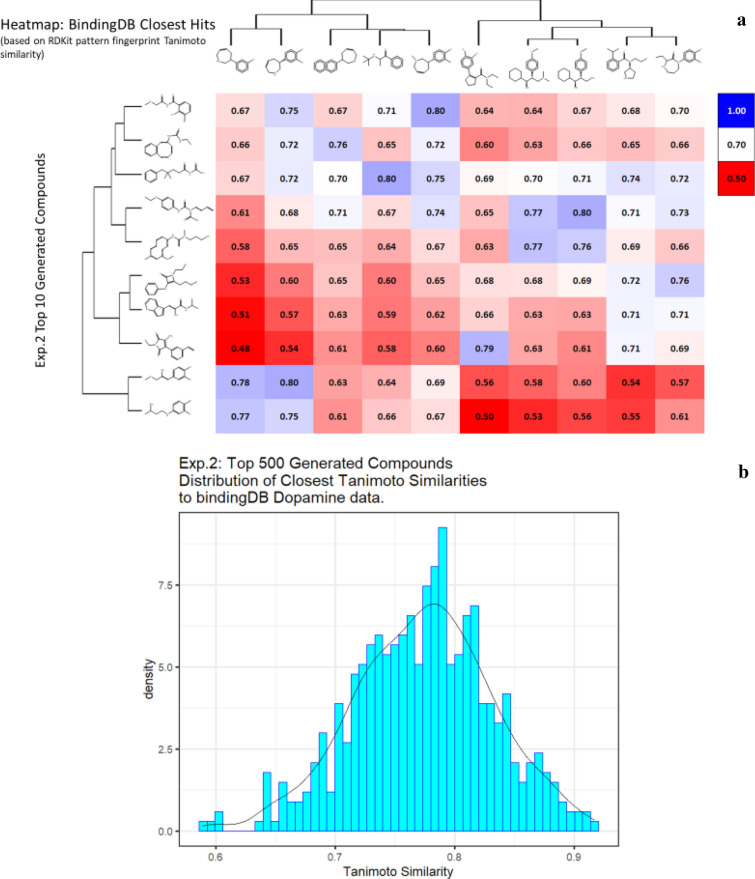
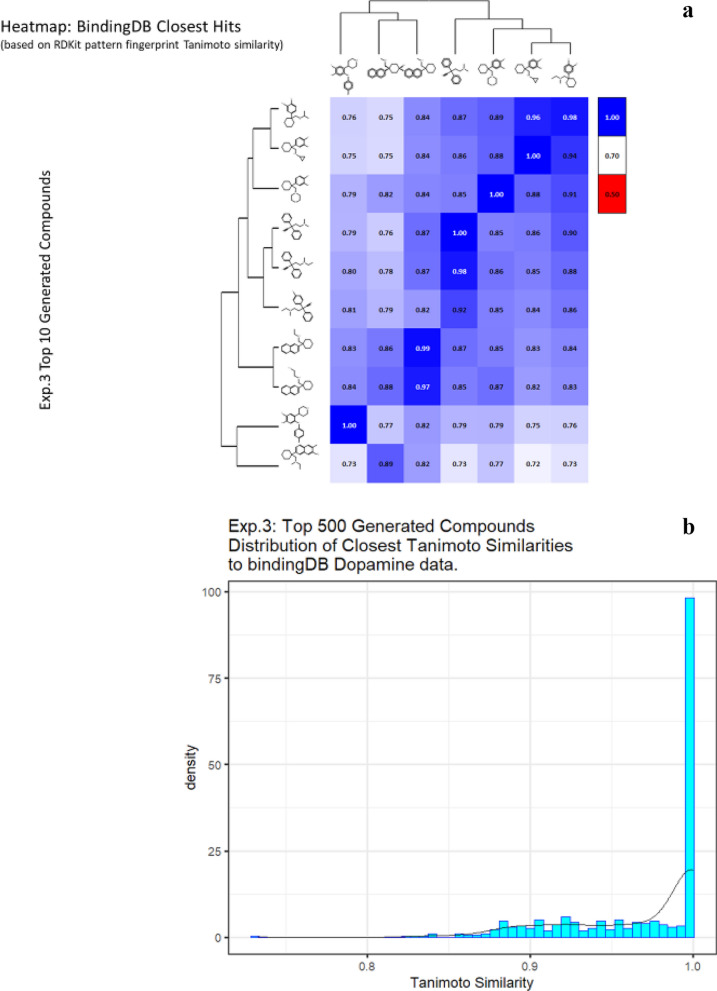
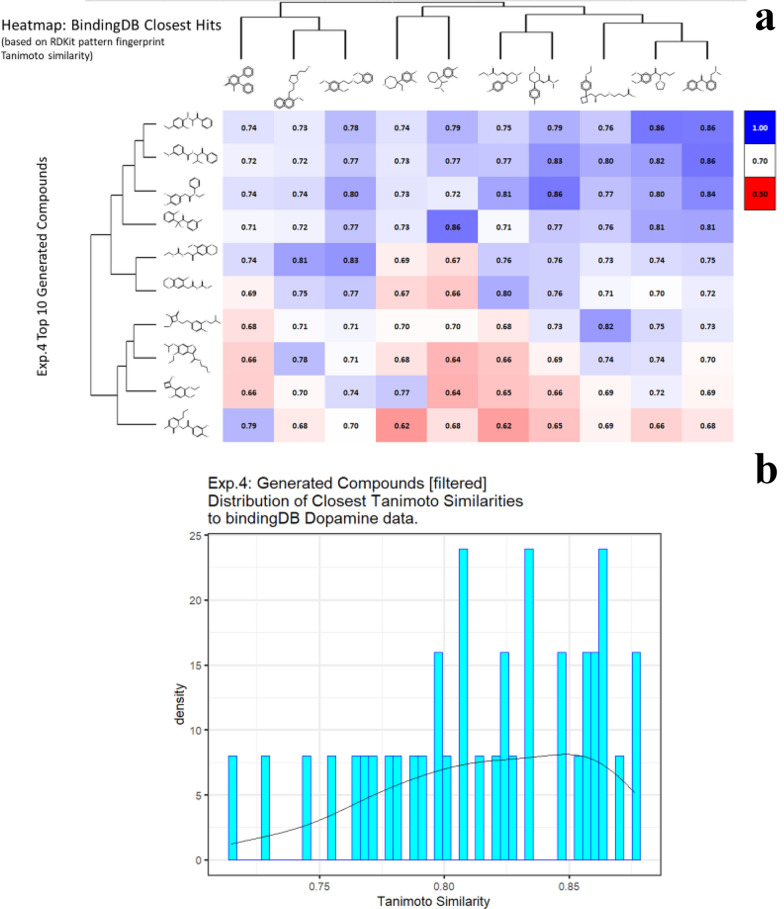
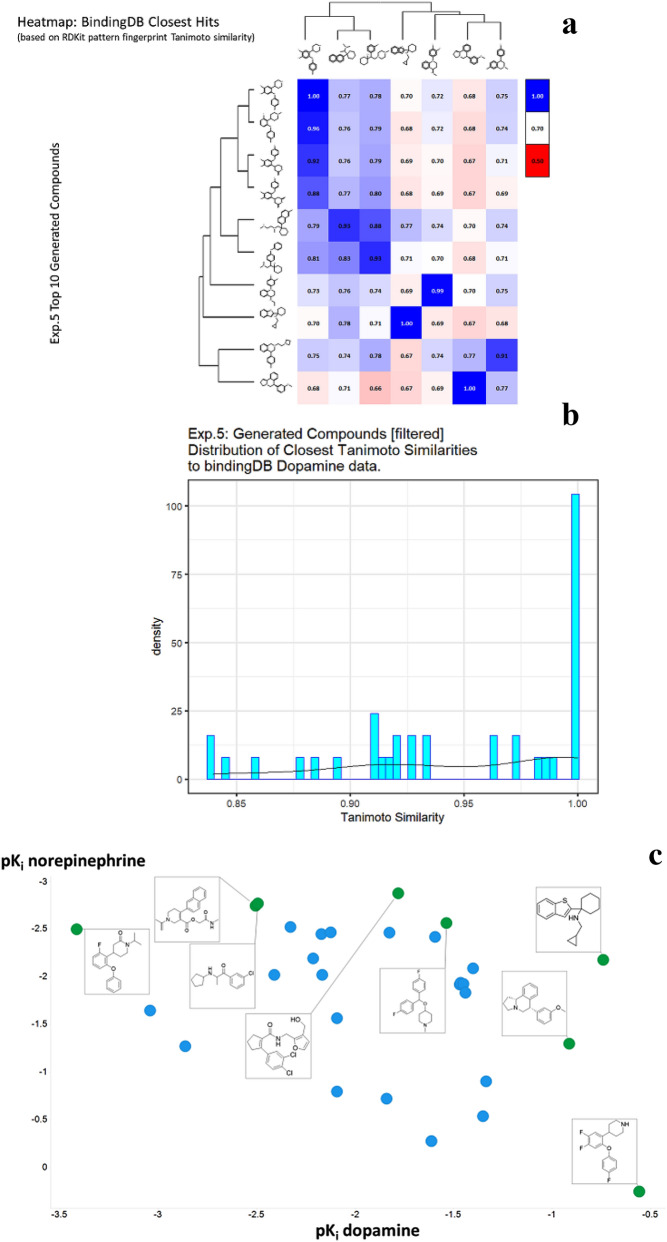
We address the problem of generating novel molecules with desired interaction properties as a multi-objective optimization problem. Interaction binding models are learned from binding data using graph convolution networks (GCNs). Since the experimentally obtained property scores are recognised as having potentially gross errors, we adopted a robust loss for the model. Combinations of these terms, including drug likeness and synthetic accessibility, are then optimized using reinforcement learning based on a graph convolution policy approach. Some of the molecules generated, while legitimate chemically, can have excellent drug-likeness scores but appear unusual. We provide an example based on the binding potency of small molecules to dopamine transporters. We extend our method successfully to use a multi-objective reward function, in this case for generating novel molecules that bind with dopamine transporters but not with those for norepinephrine. Our method should be generally applicable to the generation in silico of molecules with desirable properties.
Keywords: Cheminformatics; Deep learning; Generative methods; QSAR; Reinforcement learning.
Conflict of interest statement
The authors have no conflicts of interest to report.
Figures

Similar articles
-
Molecule generation toward target protein (SARS-CoV-2) using reinforcement learning-based graph neural network via knowledge graph.Netw Model Anal Health Inform Bioinform. 2023;12(1):13. doi: 10.1007/s13721-023-00409-2. Epub 2023 Jan 6. Netw Model Anal Health Inform Bioinform. 2023. PMID: 36627927 Free PMC article.
-
De Novo Drug Design Using Reinforcement Learning with Graph-Based Deep Generative Models.J Chem Inf Model. 2022 Oct 24;62(20):4863-4872. doi: 10.1021/acs.jcim.2c00838. Epub 2022 Oct 11. J Chem Inf Model. 2022. PMID: 36219571
-
Network-principled deep generative models for designing drug combinations as graph sets.Bioinformatics. 2020 Jul 1;36(Suppl_1):i445-i454. doi: 10.1093/bioinformatics/btaa317. Bioinformatics. 2020. PMID: 32657357 Free PMC article.
-
MGCVAE: Multi-Objective Inverse Design via Molecular Graph Conditional Variational Autoencoder.J Chem Inf Model. 2022 Jun 27;62(12):2943-2950. doi: 10.1021/acs.jcim.2c00487. Epub 2022 Jun 6. J Chem Inf Model. 2022. PMID: 35666276 Review.
-
The power of deep learning to ligand-based novel drug discovery.Expert Opin Drug Discov. 2020 Jul;15(7):755-764. doi: 10.1080/17460441.2020.1745183. Epub 2020 Mar 31. Expert Opin Drug Discov. 2020. PMID: 32228116 Review.
Cited by
-
Deep learning and generative methods in cheminformatics and chemical biology: navigating small molecule space intelligently.Biochem J. 2020 Dec 11;477(23):4559-4580. doi: 10.1042/BCJ20200781. Biochem J. 2020. PMID: 33290527 Free PMC article. Review.
-
Generative artificial intelligence based models optimization towards molecule design enhancement.J Cheminform. 2025 Aug 4;17(1):116. doi: 10.1186/s13321-025-01059-4. J Cheminform. 2025. PMID: 40759950 Free PMC article. Review.
-
Open-Source Machine Learning in Computational Chemistry.J Chem Inf Model. 2023 Aug 14;63(15):4505-4532. doi: 10.1021/acs.jcim.3c00643. Epub 2023 Jul 19. J Chem Inf Model. 2023. PMID: 37466636 Free PMC article. Review.
-
A data-driven generative strategy to avoid reward hacking in multi-objective molecular design.Nat Commun. 2025 Mar 11;16(1):2409. doi: 10.1038/s41467-025-57582-3. Nat Commun. 2025. PMID: 40069140 Free PMC article.
-
Optimizing blood-brain barrier permeation through deep reinforcement learning for de novo drug design.Bioinformatics. 2021 Jul 12;37(Suppl_1):i84-i92. doi: 10.1093/bioinformatics/btab301. Bioinformatics. 2021. PMID: 34252946 Free PMC article.
References
-
- Gómez-Bombarelli R, Aguilera-Iparraguirre J, Hirzel TD, Duvenaud D, Maclaurin D, Blood-Forsythe MA, Chae HS, Einzinger M, Ha DG, Wu T, et al. Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach. Nat Mater. 2016;15(10):1120. - PubMed
-
- Sanchez-Lengeling B, Aspuru-Guzik A. Inverse molecular design using machine learning: generative models for matter engineering. Science. 2018;361(6400):360–365. - PubMed
-
- Kadurin A, Nikolenko S, Khrabrov K, Aliper A, Zhavoronkov A. druGAN: an advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico. Mol Pharm. 2017;14(9):3098–3104. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
