

Transcription shapes genome-wide histone acetylation patterns
- PMID: 33431884
- PMCID: PMC7801501
- DOI: 10.1038/s41467-020-20543-z
Transcription shapes genome-wide histone acetylation patterns
Abstract
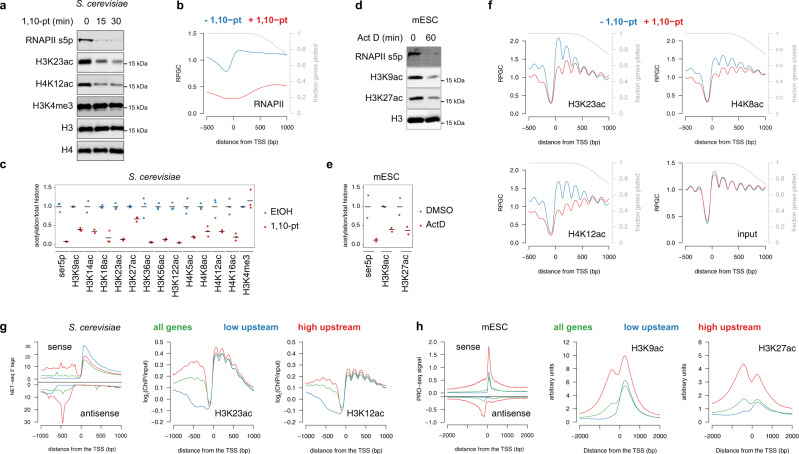
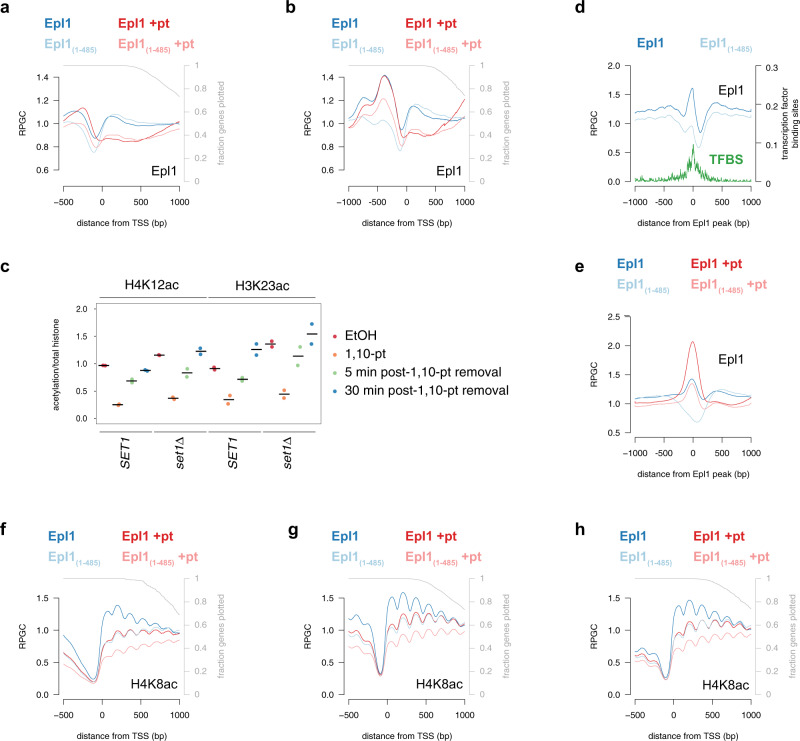
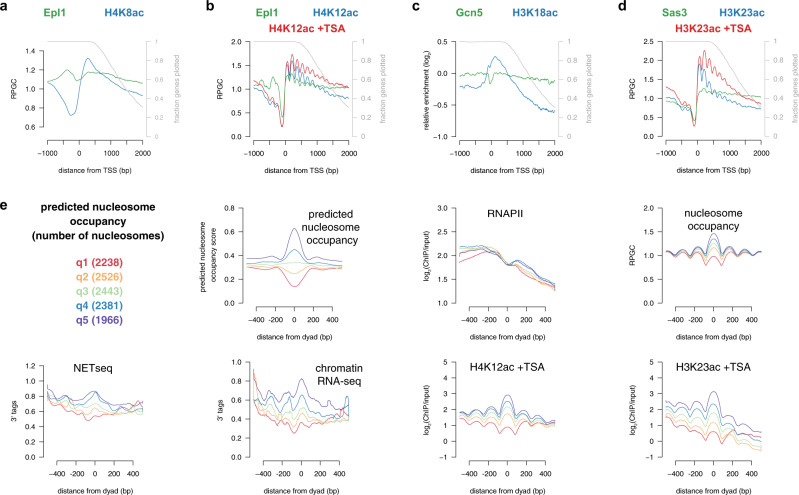
Histone acetylation is a ubiquitous hallmark of transcription, but whether the link between histone acetylation and transcription is causal or consequential has not been addressed. Using immunoblot and chromatin immunoprecipitation-sequencing in S. cerevisiae, here we show that the majority of histone acetylation is dependent on transcription. This dependency is partially explained by the requirement of RNA polymerase II (RNAPII) for the interaction of H4 histone acetyltransferases (HATs) with gene bodies. Our data also confirms the targeting of HATs by transcription activators, but interestingly, promoter-bound HATs are unable to acetylate histones in the absence of transcription. Indeed, HAT occupancy alone poorly predicts histone acetylation genome-wide, suggesting that HAT activity is regulated post-recruitment. Consistent with this, we show that histone acetylation increases at nucleosomes predicted to stall RNAPII, supporting the hypothesis that this modification is dependent on nucleosome disruption during transcription. Collectively, these data show that histone acetylation is a consequence of RNAPII promoting both the recruitment and activity of histone acetyltransferases.
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Pitfalls in using phenanthroline to study the causal relationship between promoter nucleosome acetylation and transcription.Nat Commun. 2022 Jun 29;13(1):3726. doi: 10.1038/s41467-022-30350-3. Nat Commun. 2022. PMID: 35768402 Free PMC article. No abstract available.
-
Reply to: Pitfalls in using phenanthroline to study the causal relationship between promoter nucleosome acetylation and transcription.Nat Commun. 2022 Jun 29;13(1):3725. doi: 10.1038/s41467-022-30351-2. Nat Commun. 2022. PMID: 35768425 Free PMC article. No abstract available.
References
-
- Van Holde, K. E. Chromatin. (Springer-Verlag, 1989).
-
- Rando OJ. Global patterns of histone modifications. Curr. Opin. Genet. Dev. 2007;17:94–99. - PubMed
-
- Friis RM, Schultz MC. Untargeted tail acetylation of histones in chromatin: lessons from yeast. Biochem. Cell Biol. 2009;87:107–116. - PubMed
-
- Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007;8:284–295. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
