

Exome-wide evaluation of rare coding variants using electronic health records identifies new gene-phenotype associations
- PMID: 33432171
- PMCID: PMC8775355
- DOI: 10.1038/s41591-020-1133-8
Exome-wide evaluation of rare coding variants using electronic health records identifies new gene-phenotype associations
Abstract
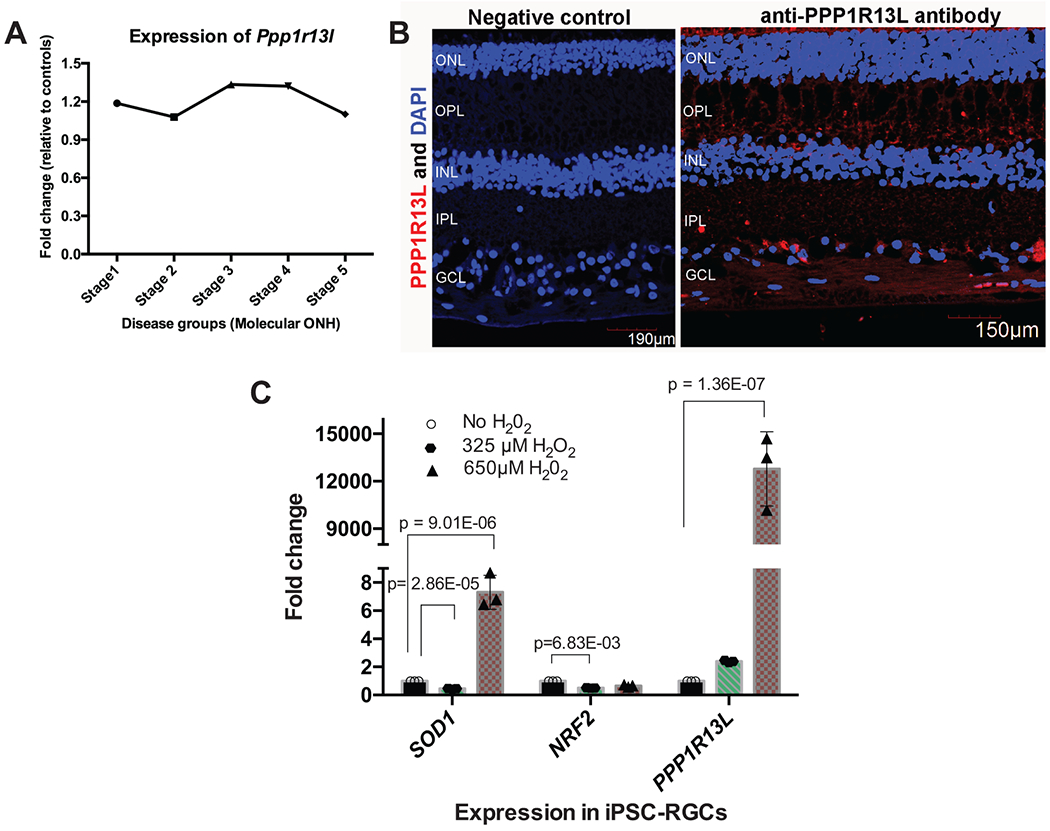
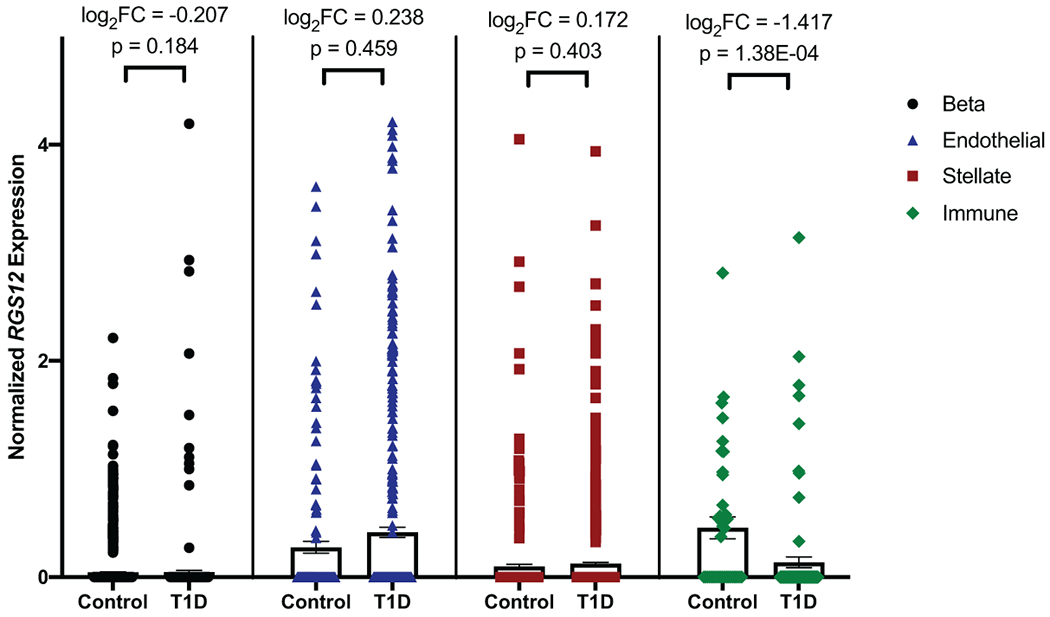
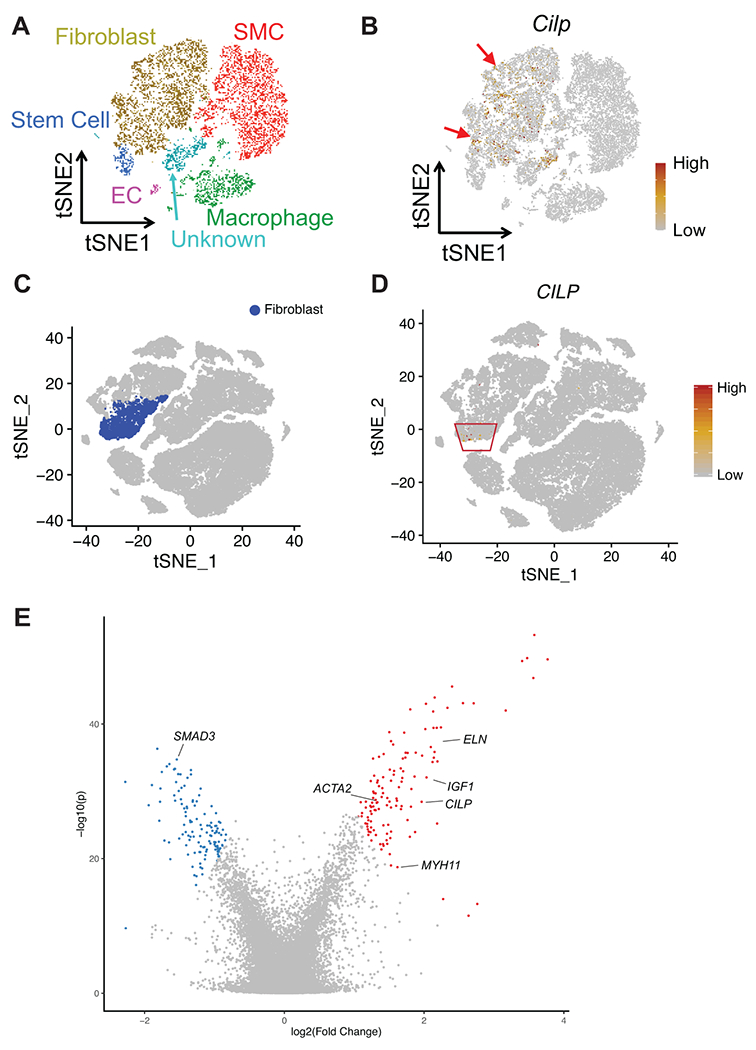
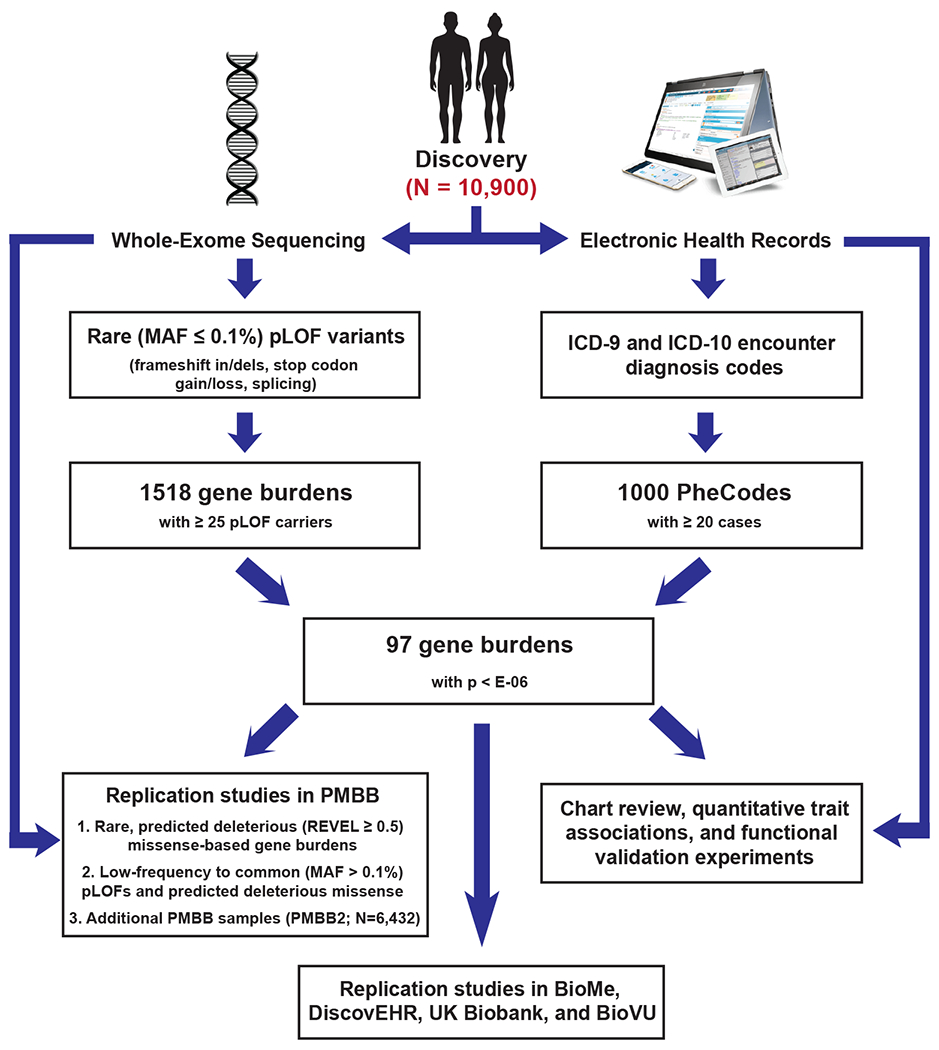
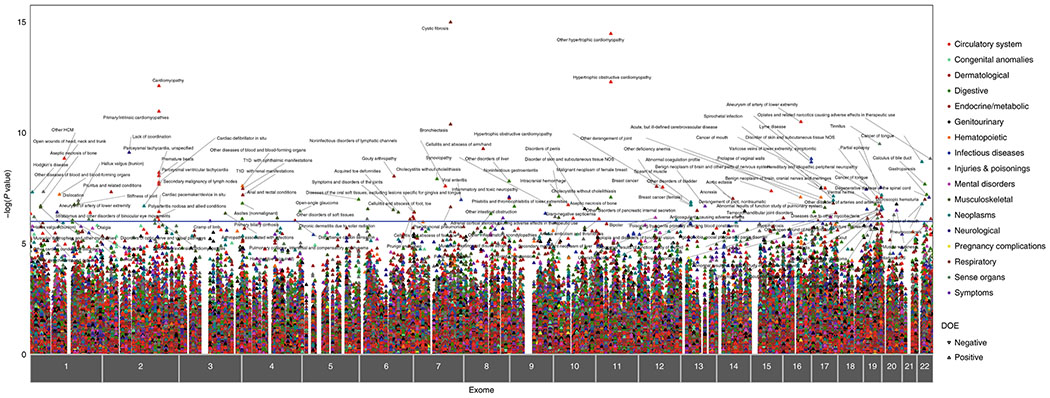
The clinical impact of rare loss-of-function variants has yet to be determined for most genes. Integration of DNA sequencing data with electronic health records (EHRs) could enhance our understanding of the contribution of rare genetic variation to human disease1. By leveraging 10,900 whole-exome sequences linked to EHR data in the Penn Medicine Biobank, we addressed the association of the cumulative effects of rare predicted loss-of-function variants for each individual gene on human disease on an exome-wide scale, as assessed using a set of diverse EHR phenotypes. After discovering 97 genes with exome-by-phenome-wide significant phenotype associations (P < 10-6), we replicated 26 of these in the Penn Medicine Biobank, as well as in three other medical biobanks and the population-based UK Biobank. Of these 26 genes, five had associations that have been previously reported and represented positive controls, whereas 21 had phenotype associations not previously reported, among which were genes implicated in glaucoma, aortic ectasia, diabetes mellitus, muscular dystrophy and hearing loss. These findings show the value of aggregating rare predicted loss-of-function variants into 'gene burdens' for identifying new gene-disease associations using EHR phenotypes in a medical biobank. We suggest that application of this approach to even larger numbers of individuals will provide the statistical power required to uncover unexplored relationships between rare genetic variation and disease phenotypes.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
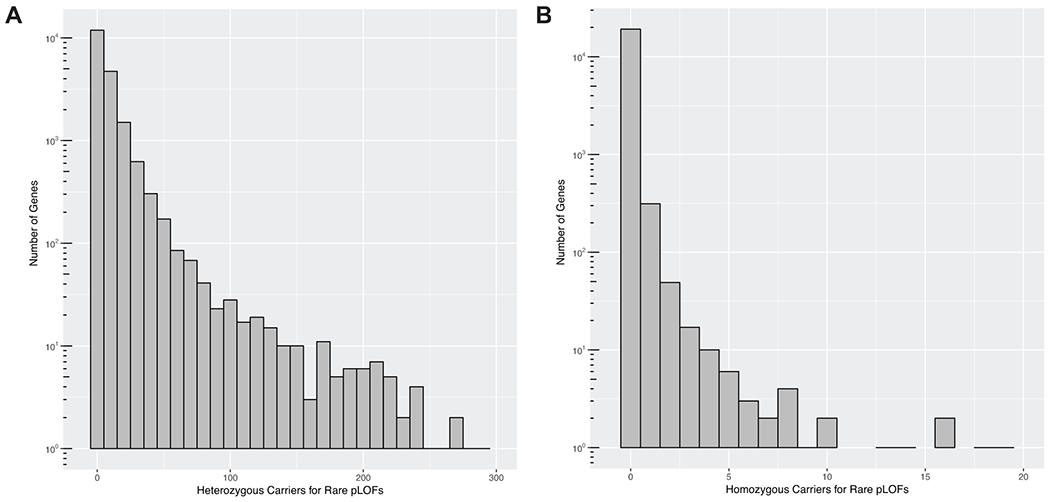
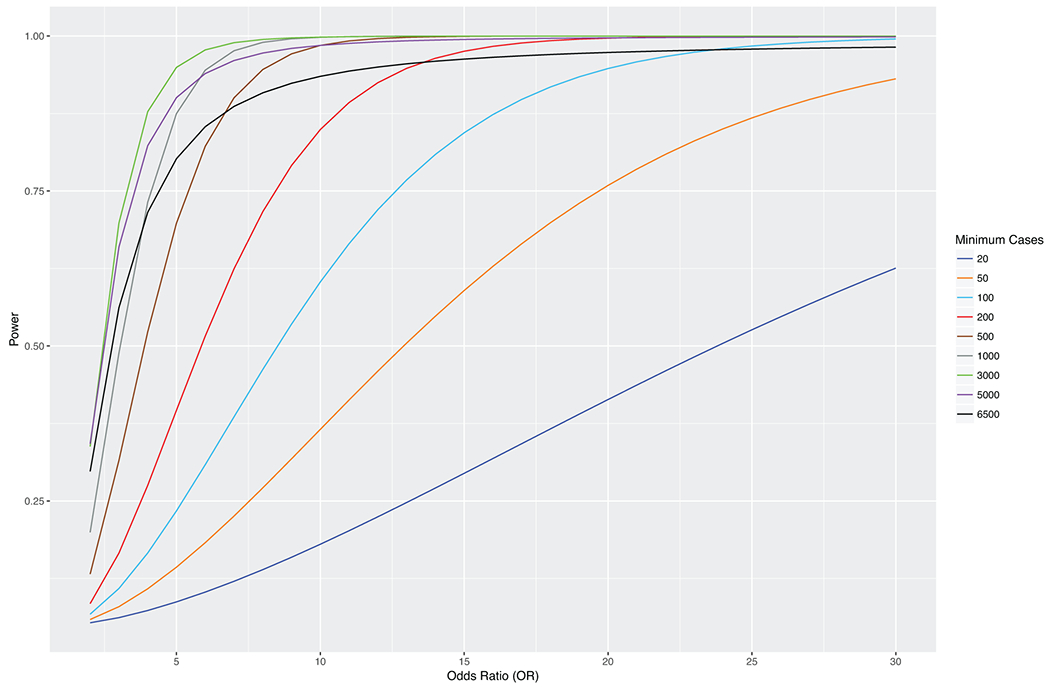
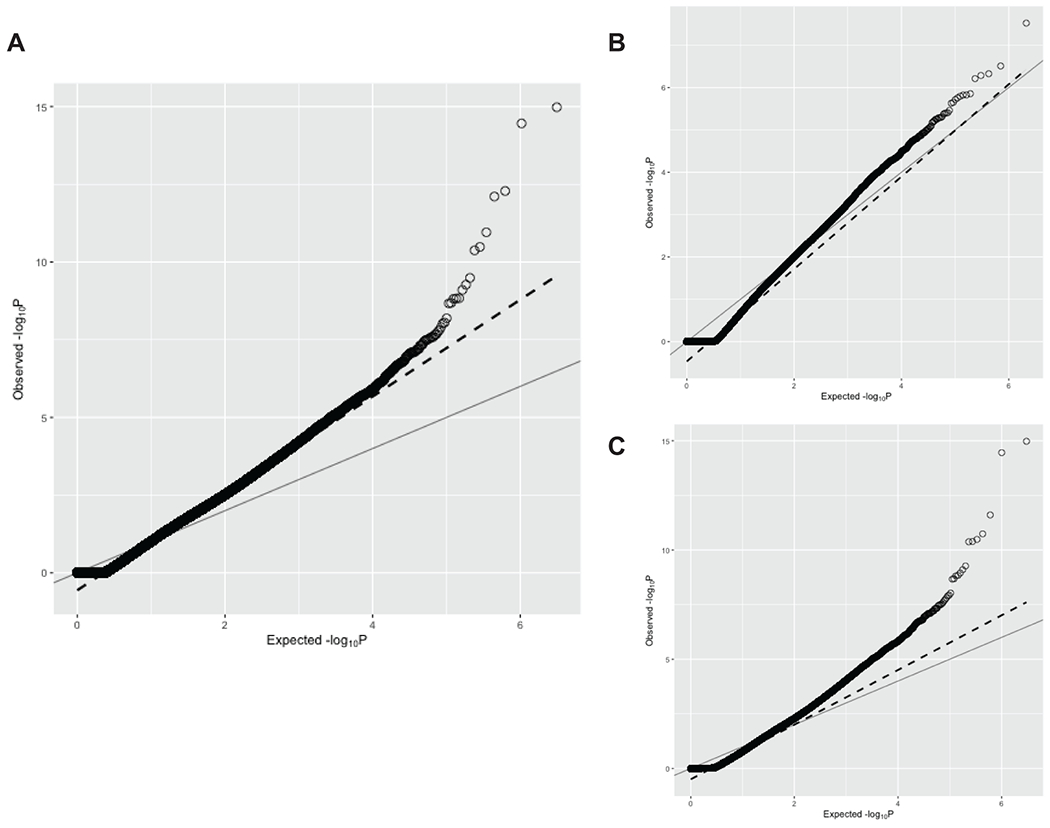
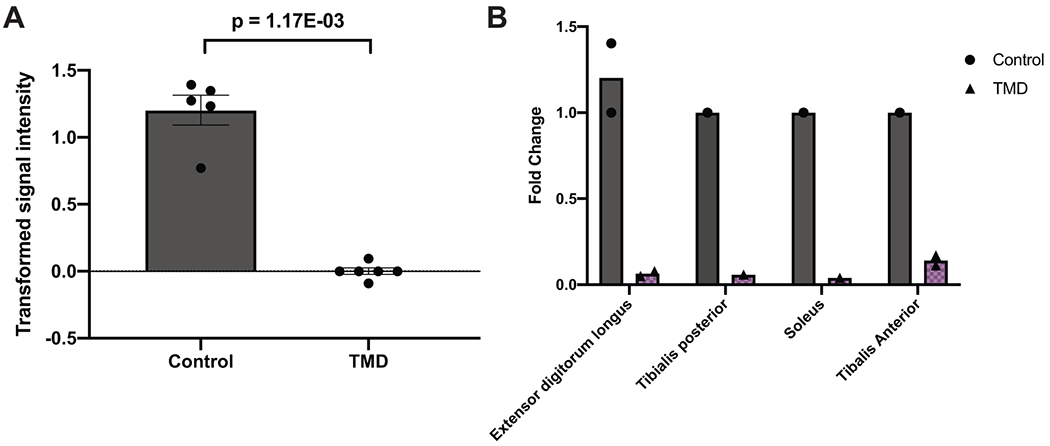
References
Methods References
Publication types
MeSH terms
Grants and funding
- T32 GM008638/GM/NIGMS NIH HHS/United States
- R01 HL143359/HL/NHLBI NIH HHS/United States
- R21EY028273-01A1/U.S. Department of Health & Human Services | NIH | National Eye Institute (NEI)/International
- UL1 TR001878/TR/NCATS NIH HHS/United States
- MC_QA137853/MRC_/Medical Research Council/United Kingdom
- R21 EY028273/EY/NEI NIH HHS/United States
- 18SFRN33960163/American Heart Association (American Heart Association, Inc.)/International
- F30HG010442/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)/International
- UL1TR001878/U.S. Department of Health & Human Services | NIH | National Center for Advancing Translational Sciences (NCATS)/International
- IK2 CX001780/CX/CSRD VA/United States
- 1R01HL143359/U.S. Department of Health & Human Services | National Institutes of Health (NIH)/International
- 18SFRN33960114/American Heart Association (American Heart Association, Inc.)/International
- F30 HG010442/HG/NHGRI NIH HHS/United States
- MC_PC_17228/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
