

Revising transcriptome assemblies with phylogenetic information
- PMID: 33434218
- PMCID: PMC7802918
- DOI: 10.1371/journal.pone.0244202
Revising transcriptome assemblies with phylogenetic information
Abstract
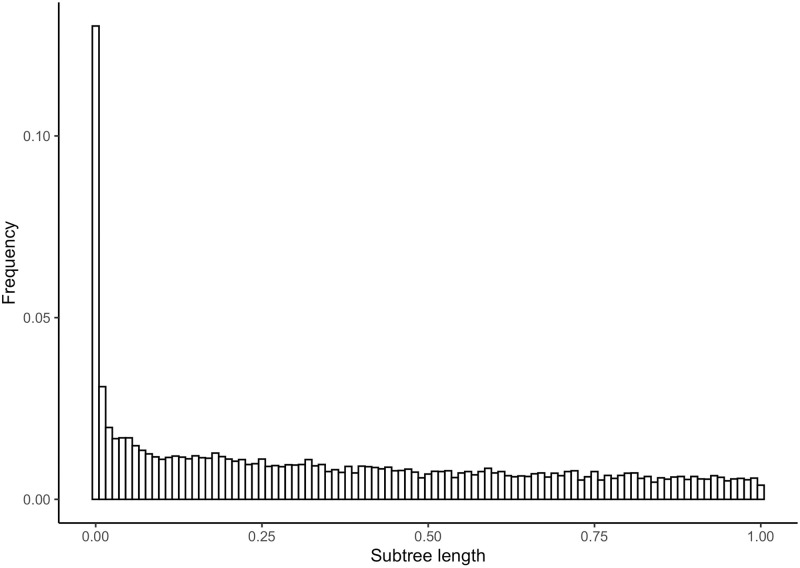
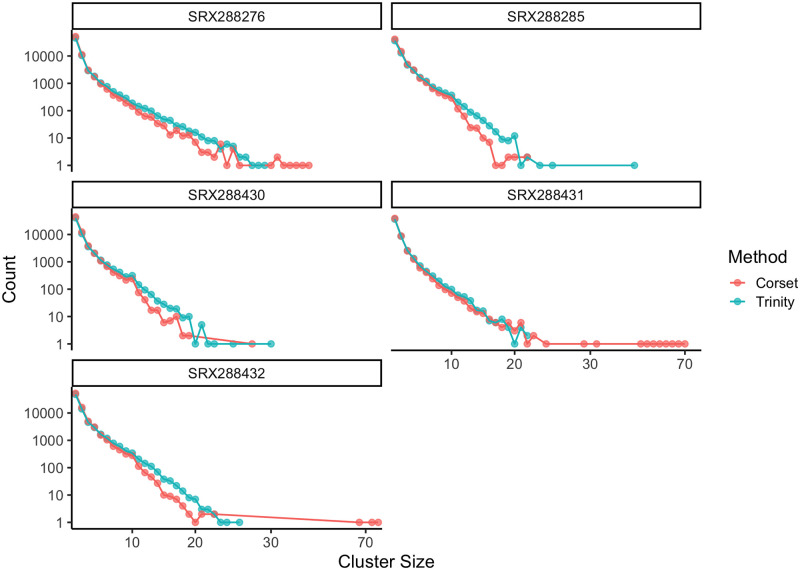
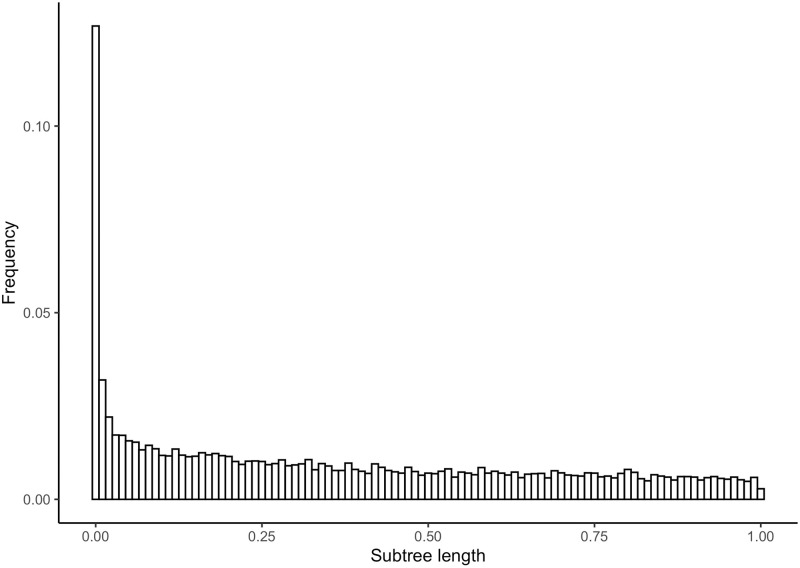
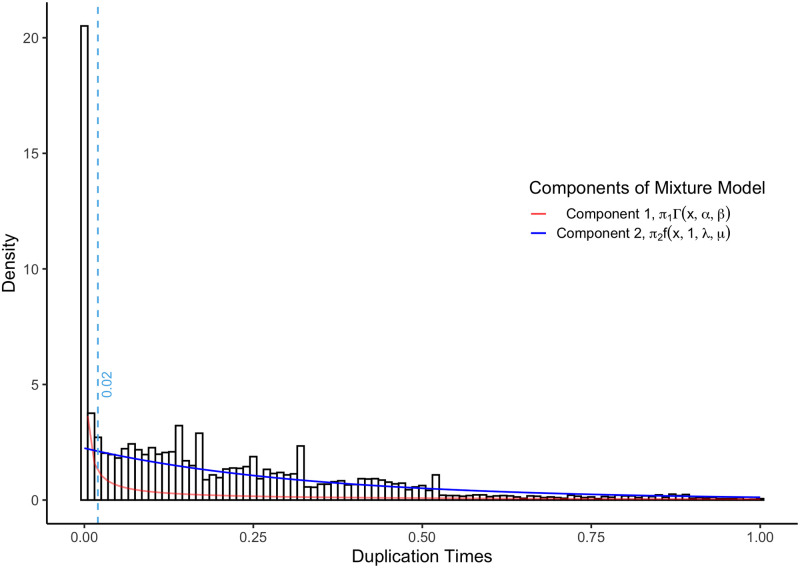
A common transcriptome assembly error is to mistake different transcripts of the same gene as transcripts from multiple closely related genes. This error is difficult to identify during assembly, but in a phylogenetic analysis such errors can be diagnosed from gene phylogenies where they appear as clades of tips from the same species with improbably short branch lengths. treeinform is a method that uses phylogenetic information across species to refine transcriptome assemblies within species. It identifies transcripts of the same gene that were incorrectly assigned to multiple genes and reassign them as transcripts of the same gene. The treeinform method is implemented in Agalma, available at https://bitbucket.org/caseywdunn/agalma, and the general approach is relevant in a variety of other contexts.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

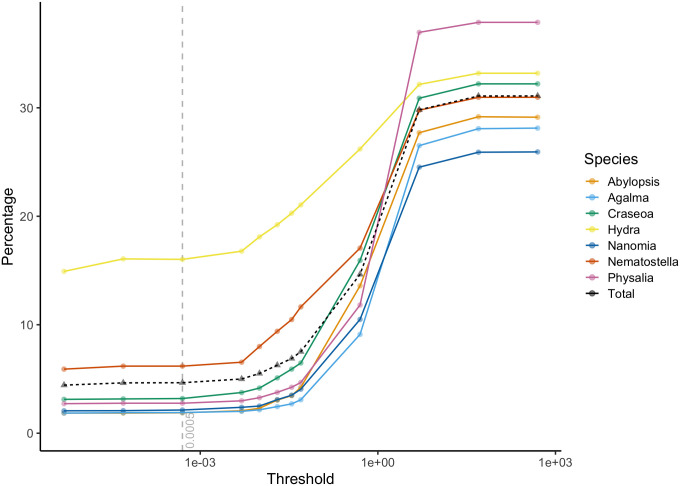
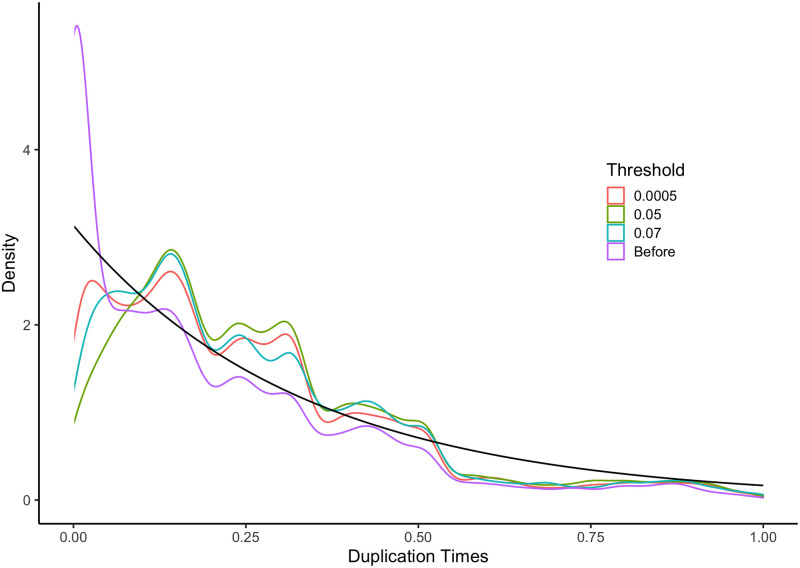
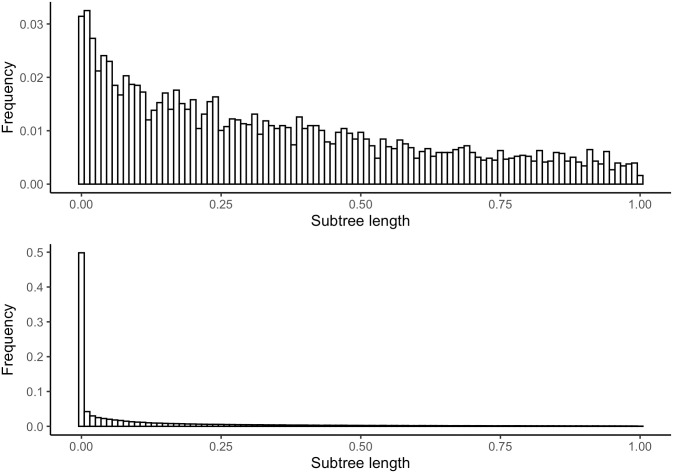
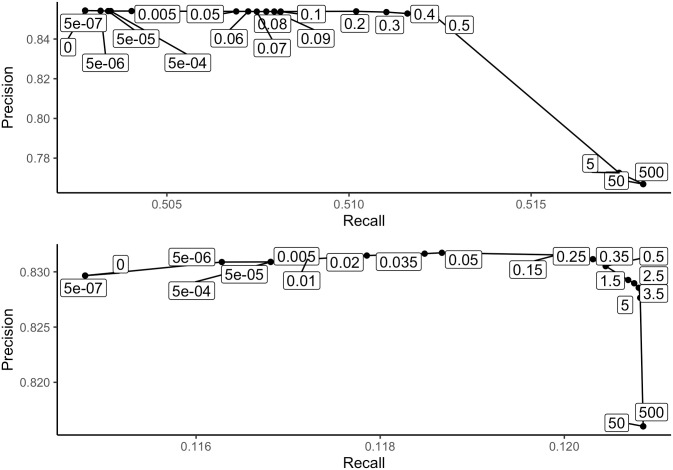
Similar articles
-
Agalma: an automated phylogenomics workflow.BMC Bioinformatics. 2013 Nov 19;14:330. doi: 10.1186/1471-2105-14-330. BMC Bioinformatics. 2013. PMID: 24252138 Free PMC article.
-
De Novo Assembly and Characterization of Four Anthozoan (Phylum Cnidaria) Transcriptomes.G3 (Bethesda). 2015 Sep 17;5(11):2441-52. doi: 10.1534/g3.115.020164. G3 (Bethesda). 2015. PMID: 26384772 Free PMC article.
-
Identification and qualification of 500 nuclear, single-copy, orthologous genes for the Eupulmonata (Gastropoda) using transcriptome sequencing and exon capture.Mol Ecol Resour. 2016 Sep;16(5):1107-23. doi: 10.1111/1755-0998.12552. Epub 2016 Aug 8. Mol Ecol Resour. 2016. PMID: 27289081
-
Molecular and developmental biology of the Cnidaria--basic aspects and phylogenetic implications.Aust J Biotechnol. 1990 Oct;4(4):241-5, 250. Aust J Biotechnol. 1990. PMID: 1369445 Review.
-
The Road To Cnidaria: History of Phylogeny of the Myxozoa.J Parasitol. 2015 Jun;101(3):269-74. doi: 10.1645/14-671.1. Epub 2015 Jan 26. J Parasitol. 2015. PMID: 25621522 Review.
Cited by
-
Phylotranscriptomics Reveals Discordance in the Phylogeny of Hawaiian Drosophila and Scaptomyza (Diptera: Drosophilidae).Mol Biol Evol. 2022 Mar 2;39(3):msac012. doi: 10.1093/molbev/msac012. Mol Biol Evol. 2022. PMID: 35048974 Free PMC article.
-
Improved phylogenetic resolution within Siphonophora (Cnidaria) with implications for trait evolution.Mol Phylogenet Evol. 2018 Oct;127:823-833. doi: 10.1016/j.ympev.2018.06.030. Epub 2018 Jun 22. Mol Phylogenet Evol. 2018. PMID: 29940256 Free PMC article.
-
Phylogenomic analyses of echinoid diversification prompt a re-evaluation of their fossil record.Elife. 2022 Mar 22;11:e72460. doi: 10.7554/eLife.72460. Elife. 2022. PMID: 35315317 Free PMC article.
-
Automatic identification and annotation of MYB gene family members in plants.BMC Genomics. 2022 Mar 19;23(1):220. doi: 10.1186/s12864-022-08452-5. BMC Genomics. 2022. PMID: 35305581 Free PMC article.
-
Comparative Genomics Uncovers the Evolutionary Dynamics of Detoxification and Insecticide Target Genes Across 11 Phlebotomine Sand Flies.Genome Biol Evol. 2024 Sep 3;16(9):evae186. doi: 10.1093/gbe/evae186. Genome Biol Evol. 2024. PMID: 39224065 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
