

A modular master regulator landscape controls cancer transcriptional identity
- PMID: 33434495
- PMCID: PMC8103356
- DOI: 10.1016/j.cell.2020.11.045
A modular master regulator landscape controls cancer transcriptional identity
Abstract
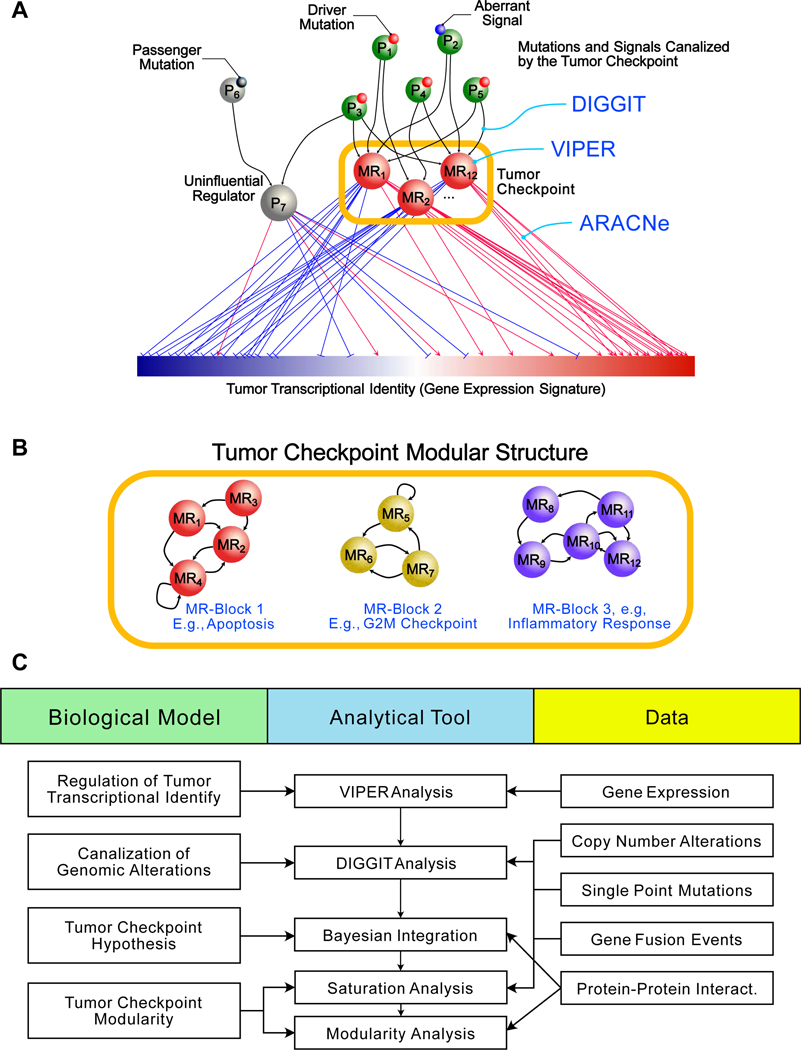
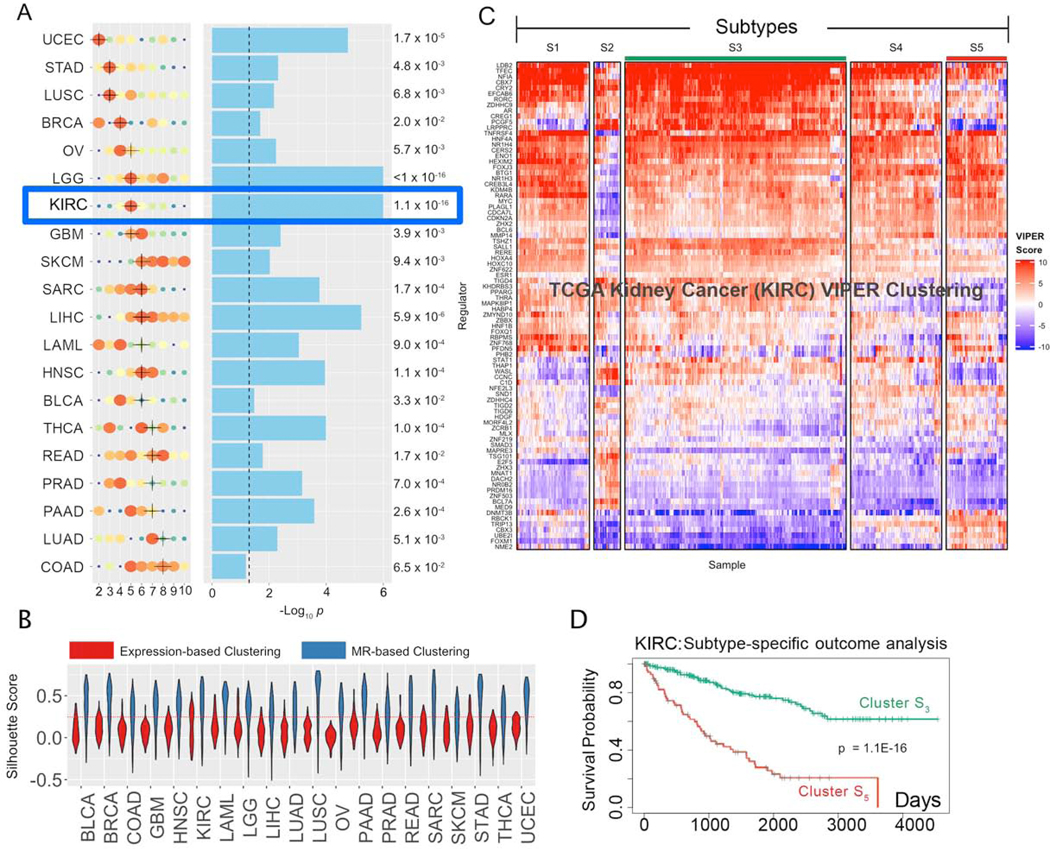
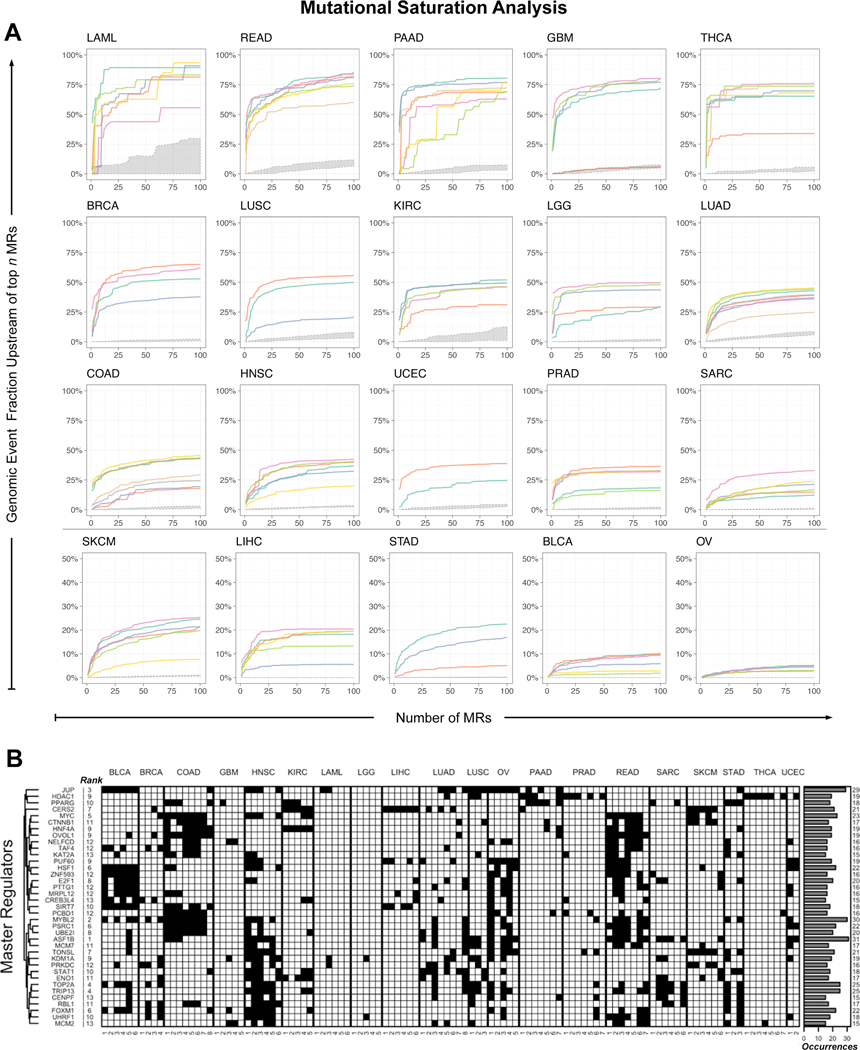
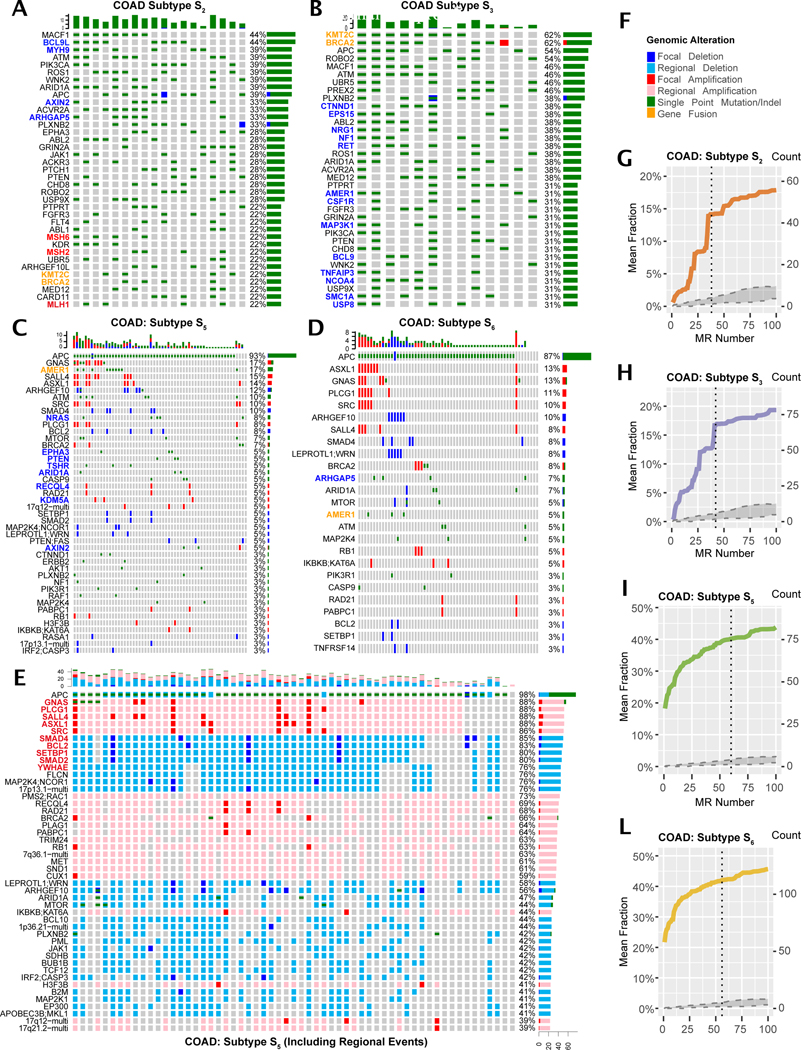
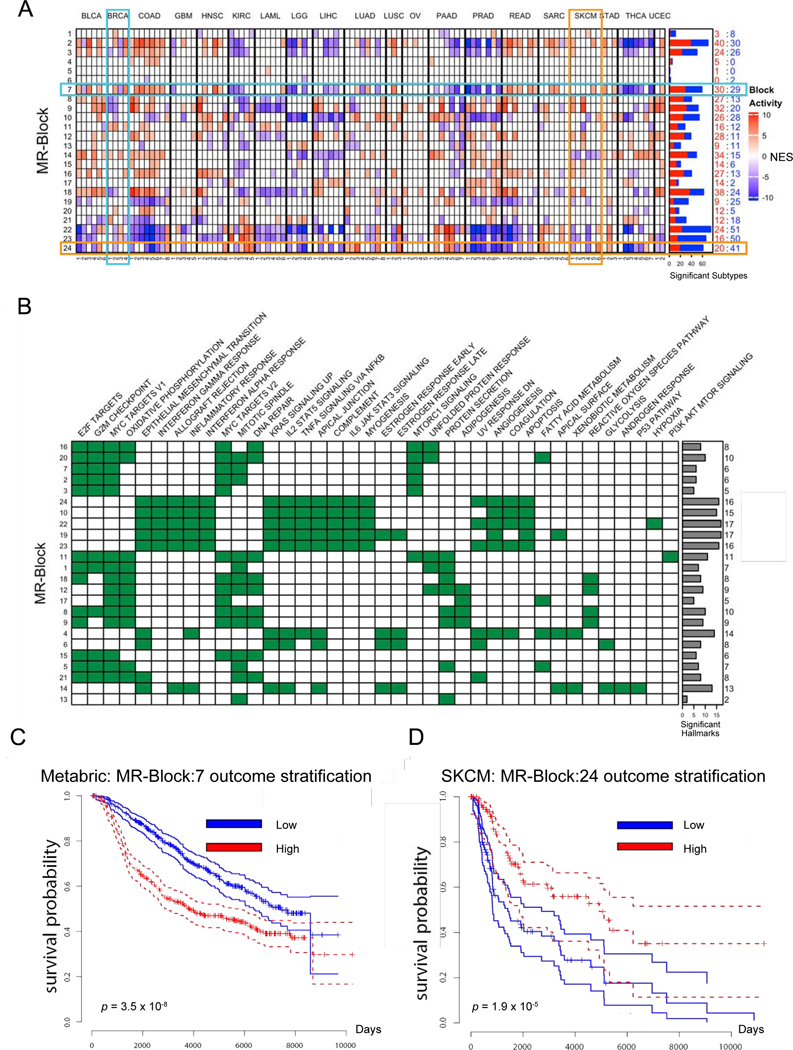
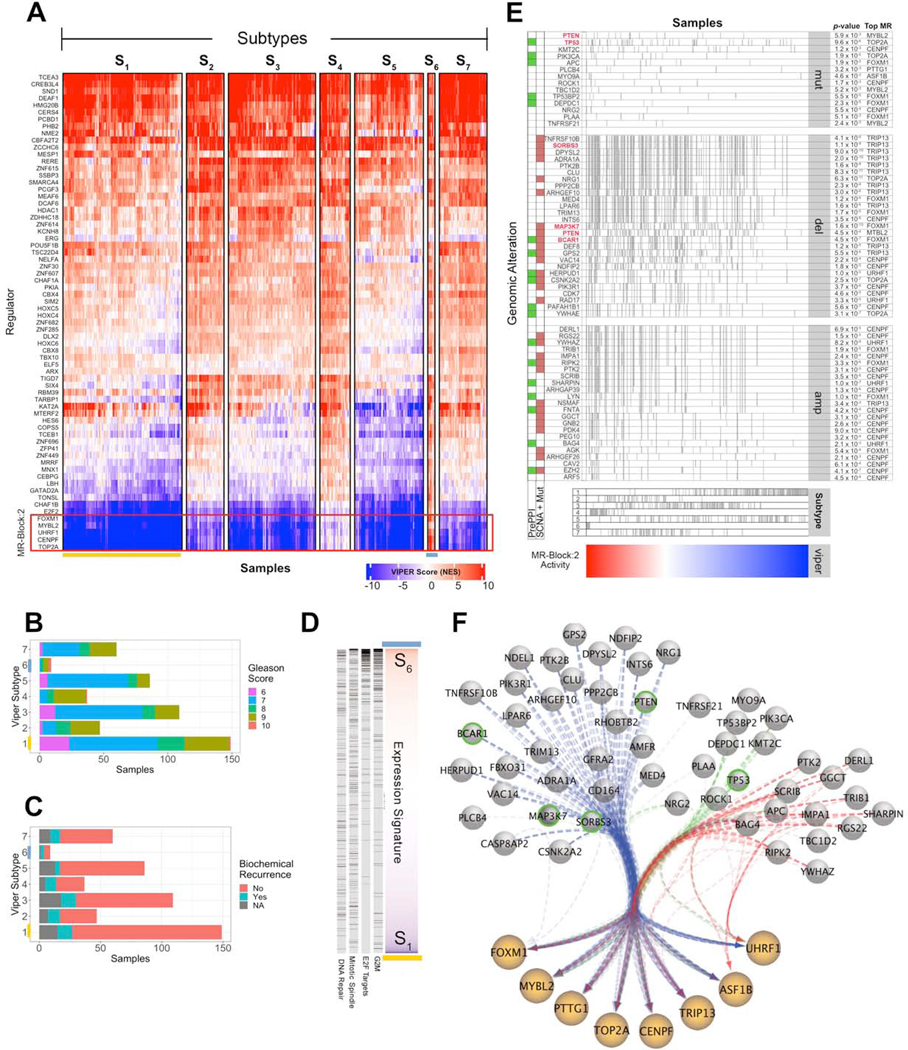
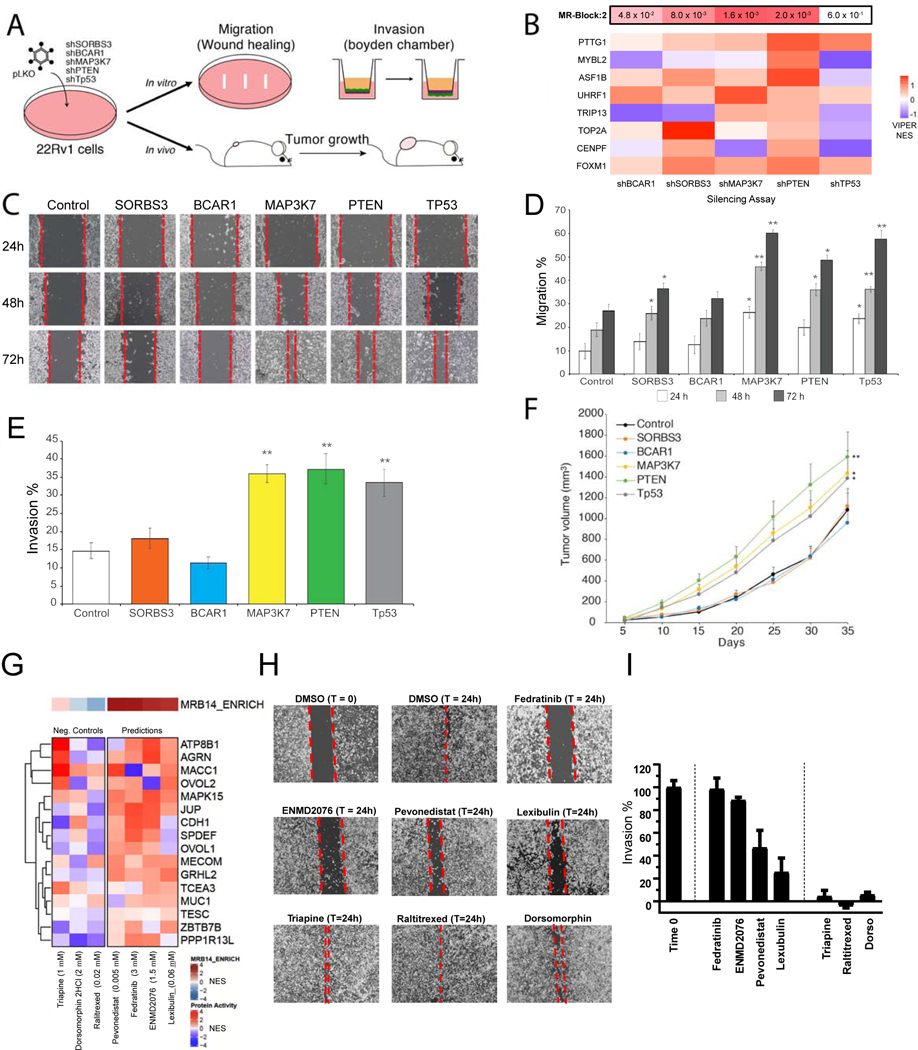
Despite considerable efforts, the mechanisms linking genomic alterations to the transcriptional identity of cancer cells remain elusive. Integrative genomic analysis, using a network-based approach, identified 407 master regulator (MR) proteins responsible for canalizing the genetics of individual samples from 20 cohorts in The Cancer Genome Atlas (TCGA) into 112 transcriptionally distinct tumor subtypes. MR proteins could be further organized into 24 pan-cancer, master regulator block modules (MRBs), each regulating key cancer hallmarks and predictive of patient outcome in multiple cohorts. Of all somatic alterations detected in each individual sample, >50% were predicted to induce aberrant MR activity, yielding insight into mechanisms linking tumor genetics and transcriptional identity and establishing non-oncogene dependencies. Genetic and pharmacological validation assays confirmed the predicted effect of upstream mutations and MR activity on downstream cellular identity and phenotype. Thus, co-analysis of mutational and gene expression profiles identified elusive subtypes and provided testable hypothesis for mechanisms mediating the effect of genetic alterations.
Keywords: cancer genetics; cancer systems biology; genomic alteration; integrative genomics; multiomics; network analysis; pan-cancer analysis; transcriptional regulation.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.C. is founder, equity holder, consultant, and director of DarwinHealth Inc., a company that has licensed some of the algorithms used in this manuscript from Columbia University. M.J.A. is Chief Scientific Officer and equity holder at DarwinHealth, Inc. Patent 10,790,040, titled “Virtual Inference of Protein Activity by Regulon Analysis” has issued on Sept. 29, 2020 related to the VIPER method. Columbia University is also an equity holder in DarwinHealth Inc.
Figures
References
-
- Aytes A, Mitrofanova A, Lefebvre C, Alvarez MJ, Castillo-Martin M, Zheng T, Eastham JA, Gopalan A, Pienta KJ, Shen MM, et al. (2014). Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer Cell 25, 638–651. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
