

A microbial metabolite remodels the gut-liver axis following bariatric surgery
- PMID: 33434516
- PMCID: PMC7954942
- DOI: 10.1016/j.chom.2020.12.004
A microbial metabolite remodels the gut-liver axis following bariatric surgery
Abstract
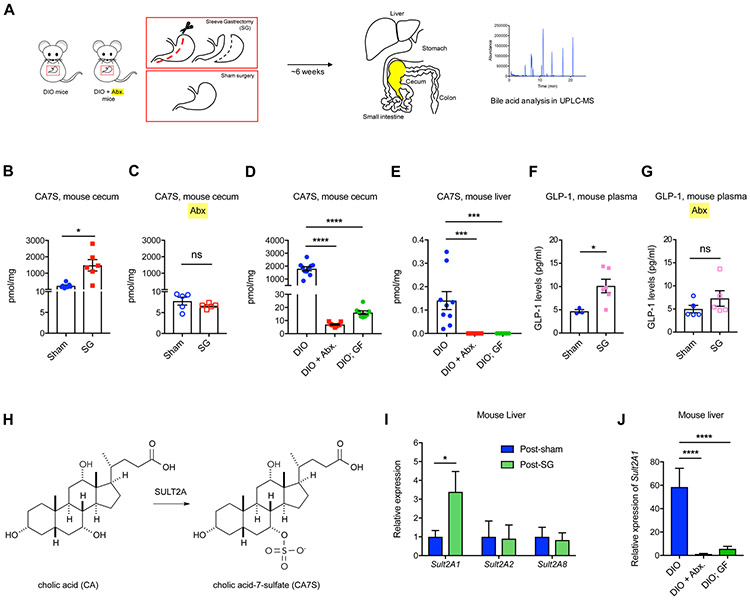
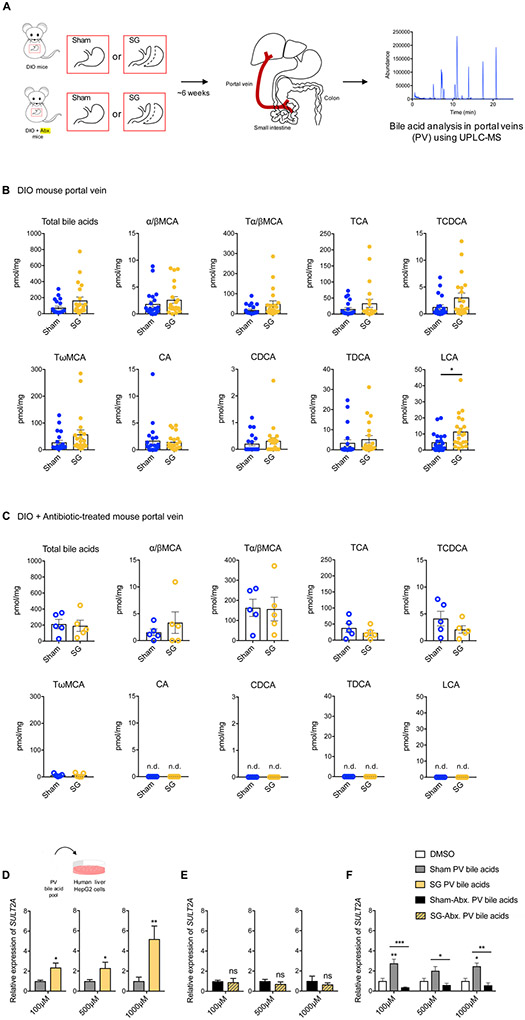
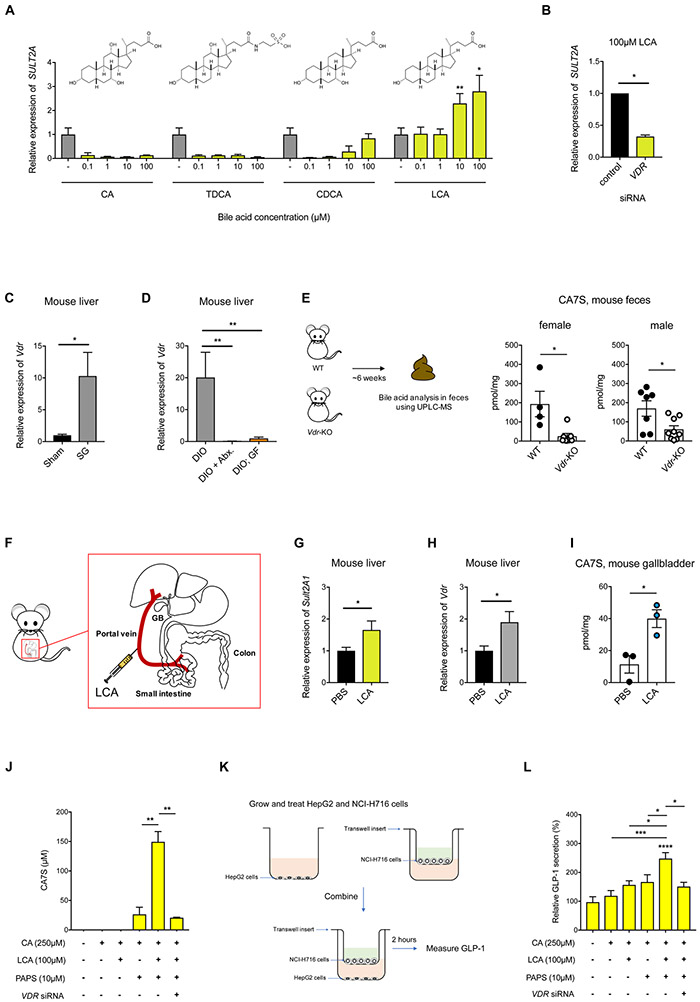
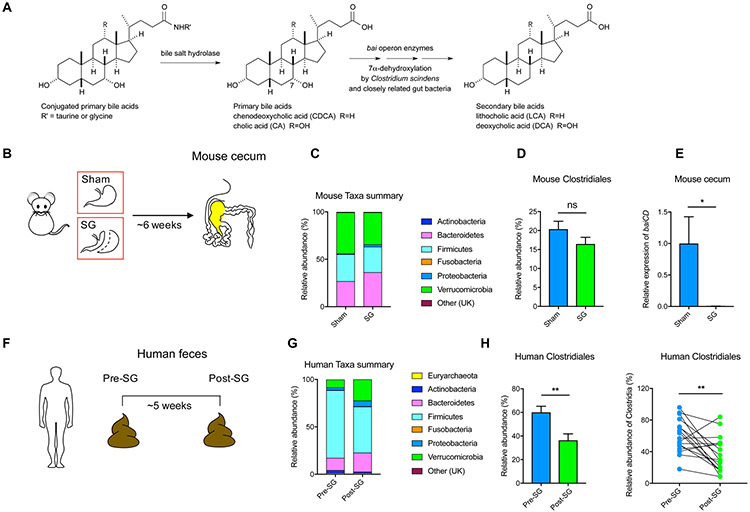
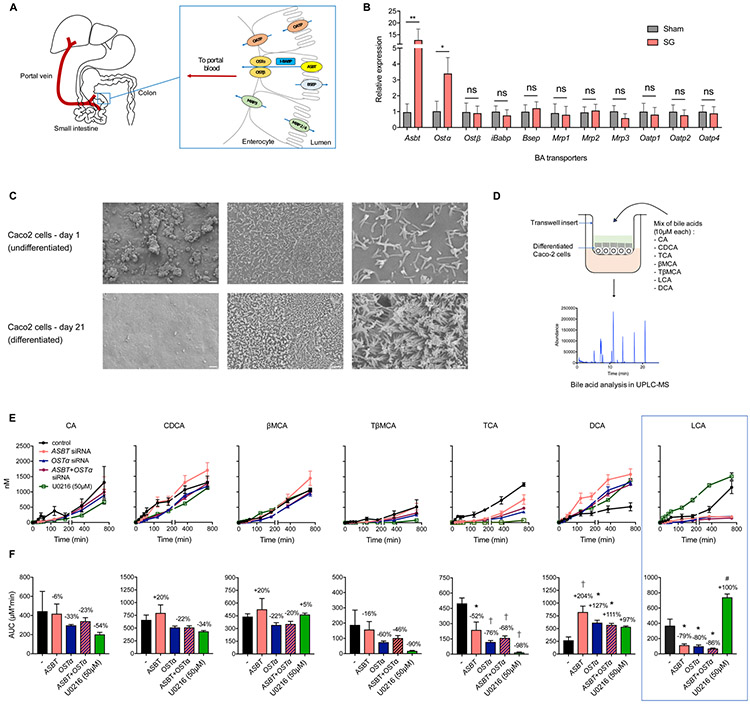
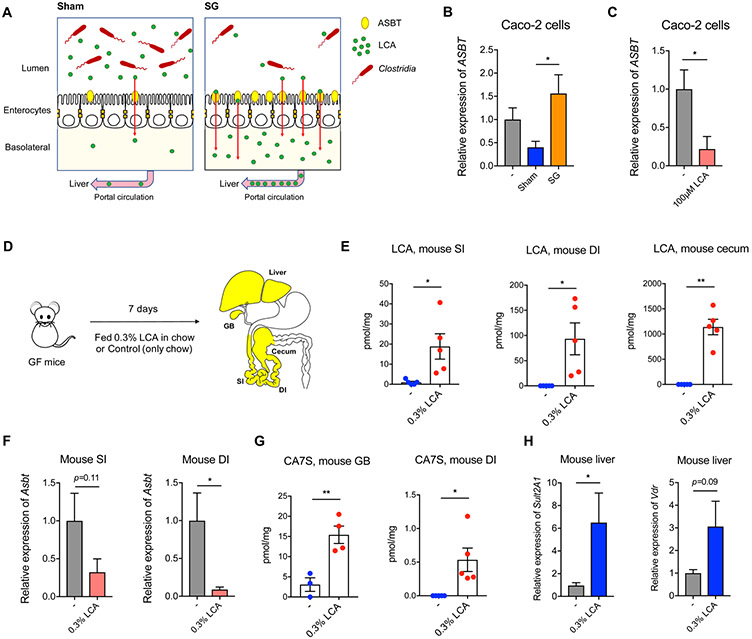
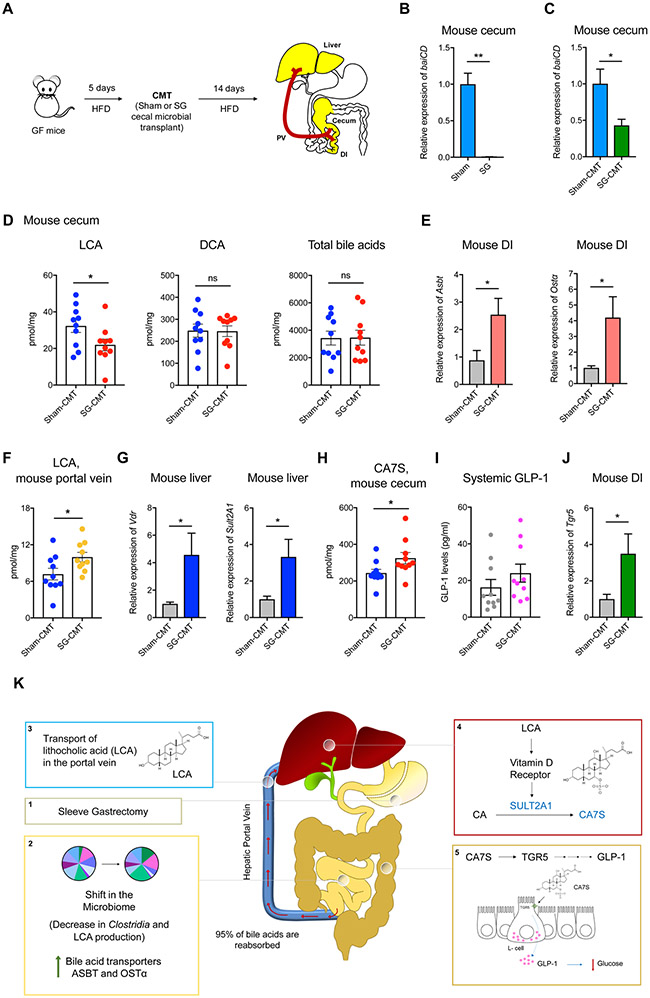
Bariatric surgery is the most effective treatment for type 2 diabetes and is associated with changes in gut metabolites. Previous work uncovered a gut-restricted TGR5 agonist with anti-diabetic properties-cholic acid-7-sulfate (CA7S)-that is elevated following sleeve gastrectomy (SG). Here, we elucidate a microbiome-dependent pathway by which SG increases CA7S production. We show that a microbial metabolite, lithocholic acid (LCA), is increased in murine portal veins post-SG and by activating the vitamin D receptor, induces hepatic mSult2A1/hSULT2A expression to drive CA7S production. An SG-induced shift in the microbiome increases gut expression of the bile acid transporters Asbt and Ostα, which in turn facilitate selective transport of LCA across the gut epithelium. Cecal microbiota transplant from SG animals is sufficient to recreate the pathway in germ-free (GF) animals. Activation of this gut-liver pathway leads to CA7S synthesis and GLP-1 secretion, causally connecting a microbial metabolite with the improvement of diabetic phenotypes.
Keywords: bariatric surgery; bile acids; gut-liver axis; microbiome.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests CA7S is a subject of patents held by HMS and BWH on which S.N.C., D.A.H., E.G.S., and A.S.D. are inventors. A.S.D. is a consultant for Takeda Pharmaceuticals and HP Hood. E.G.S. is a consultant for Vicarious Surgical, Inc. and was previously on the scientific advisory board of Kitotech, Inc.
Figures
Comment in
-
Bariatric surgery stirs symbionts to counteract diabesity by CA(7)Sting a liver-generated bile acid into the mix.Cell Host Microbe. 2021 Mar 10;29(3):320-322. doi: 10.1016/j.chom.2021.02.011. Cell Host Microbe. 2021. PMID: 33705701
References
-
- Abbasi J (2017). Unveiling the "Magic" of Diabetes Remission After Weight-Loss Surgery. JAMA 317, 571–574. - PubMed
-
- Alnouti Y (2009). Bile Acid sulfation: a pathway of bile acid elimination and detoxification. Toxicol Sci 108, 225–246. - PubMed
-
- Besnard P, Landrier JF, Grober J, and Niot I (2004). [Is the ileal bile acid-binding protein (I-BABP) gene involved in cholesterol homeostasis?]. Med Sci (Paris) 20, 73–77. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
