

Transcription Inhibitors with XRE DNA-Binding and Cupin Signal-Sensing Domains Drive Metabolic Diversification in Pseudomonas
- PMID: 33436508
- PMCID: PMC7901475
- DOI: 10.1128/mSystems.00753-20
Transcription Inhibitors with XRE DNA-Binding and Cupin Signal-Sensing Domains Drive Metabolic Diversification in Pseudomonas
Abstract
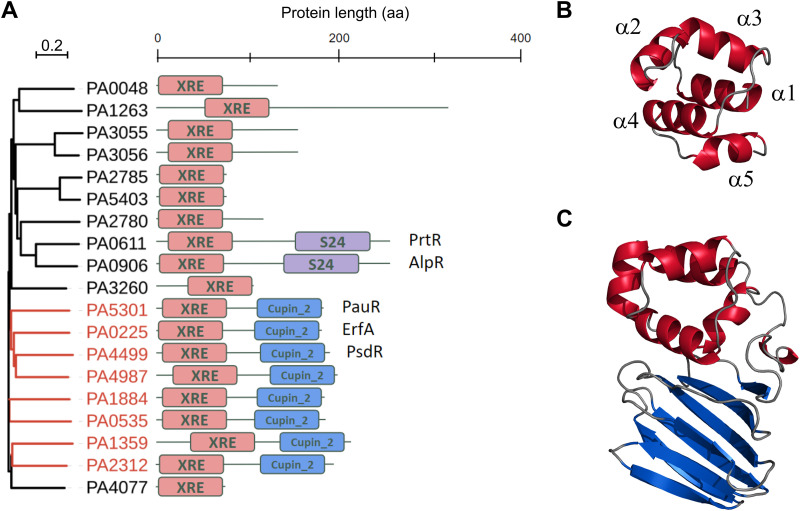
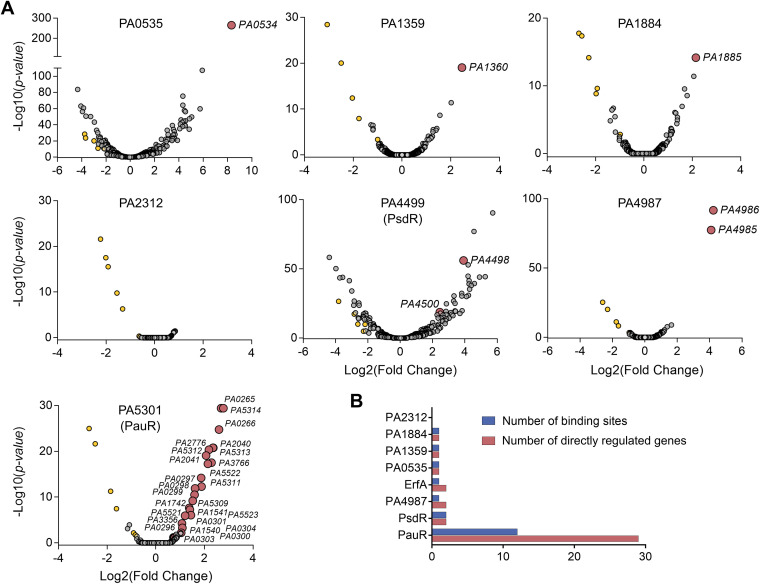
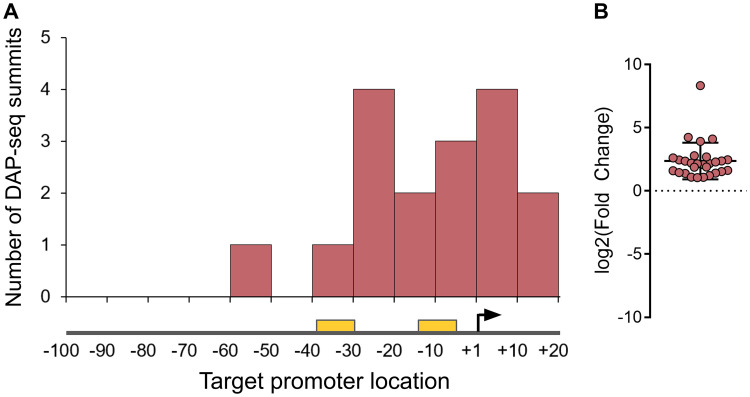
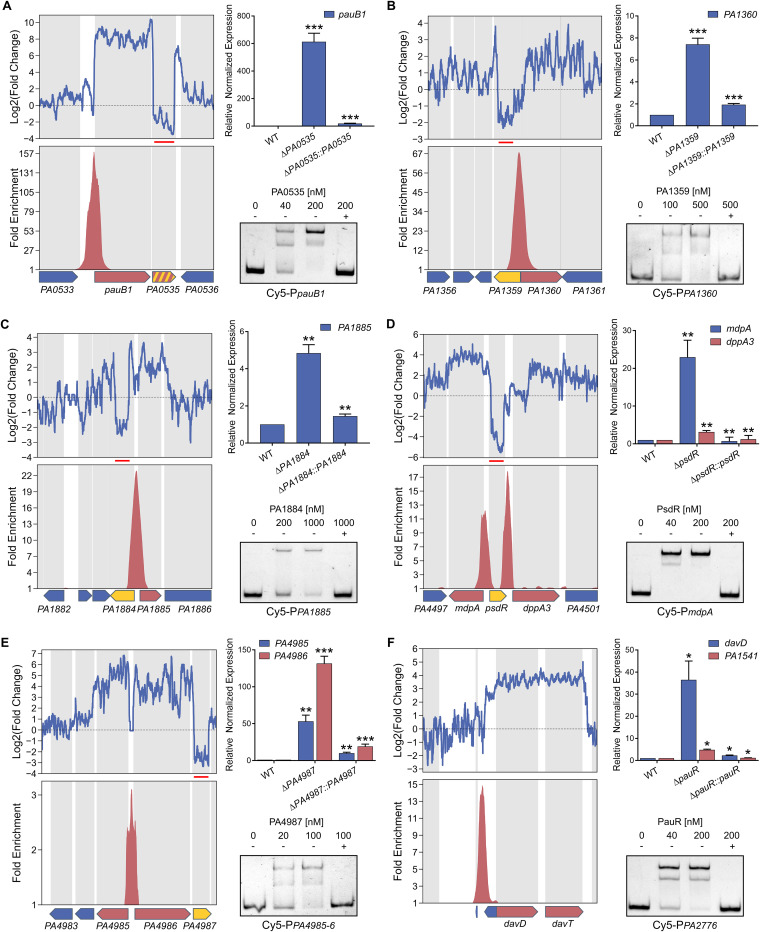
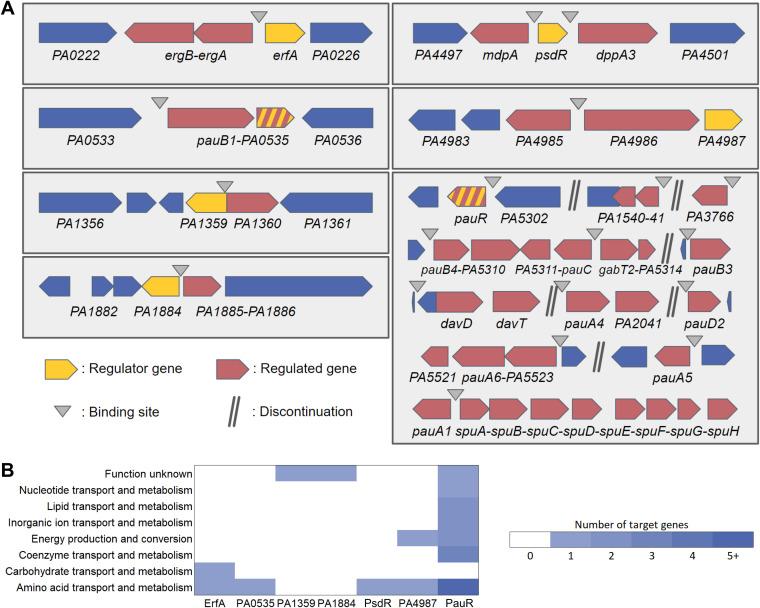
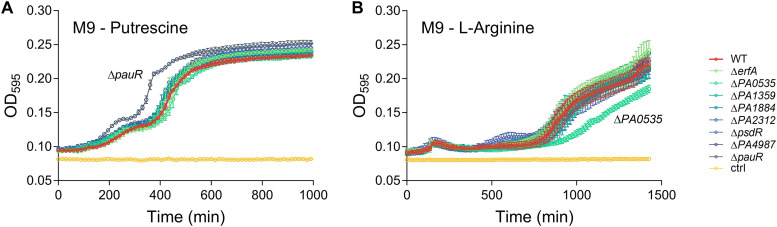
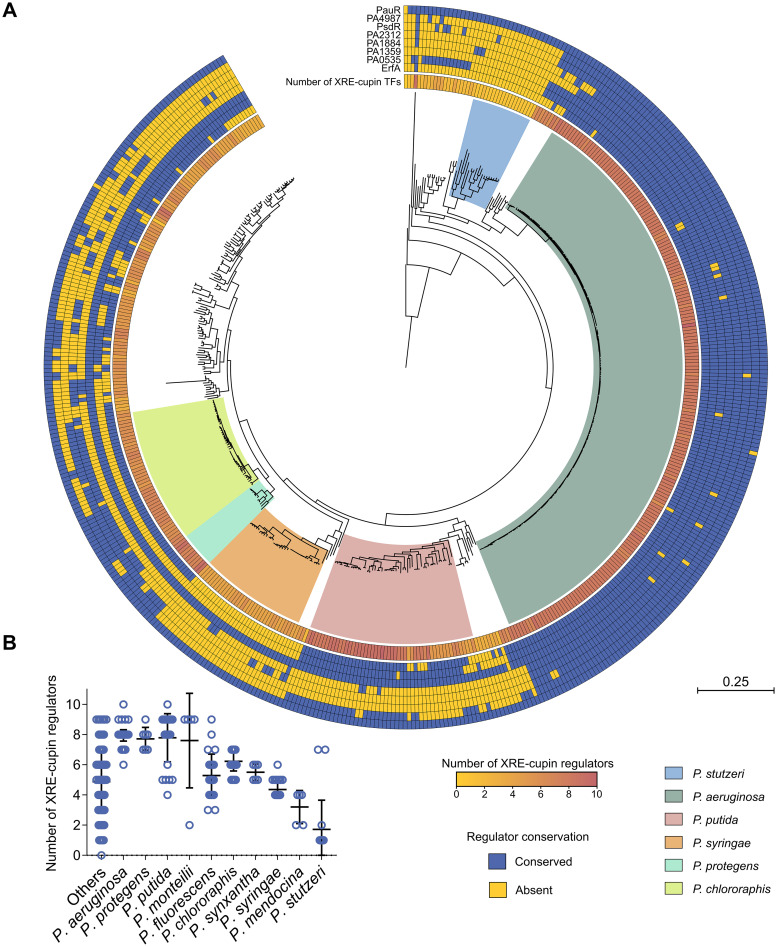
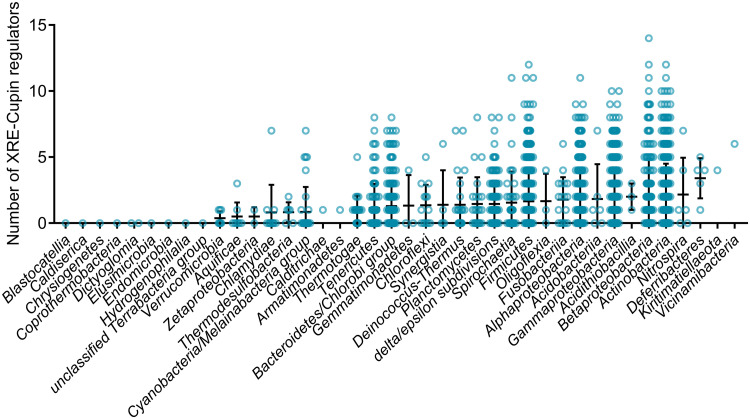
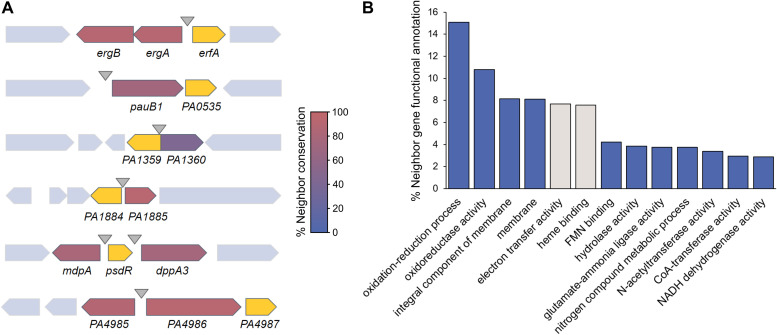
Transcription factors (TFs) are instrumental in the bacterial response to new environmental conditions. They can act as direct signal sensors and subsequently induce changes in gene expression leading to physiological adaptation. Here, by combining transcriptome sequencing (RNA-seq) and cistrome determination (DAP-seq), we studied a family of eight TFs in Pseudomonas aeruginosa This family, encompassing TFs with XRE-like DNA-binding and cupin signal-sensing domains, includes the metabolic regulators ErfA, PsdR, and PauR and five so-far-unstudied TFs. The genome-wide delineation of their regulons identified 39 regulatory interactions with genes mostly involved in metabolism. We found that the XRE-cupin TFs are inhibitors of their neighboring genes, forming local, functional units encoding proteins with functions in condition-specific metabolic pathways. Growth phenotypes of isogenic mutants highlighted new roles for PauR and PA0535 in polyamines and arginine metabolism. The phylogenetic analysis of this family of regulators across the bacterial kingdom revealed a wide diversity of such metabolic regulatory modules and identified species with potentially higher metabolic versatility. Numerous genes encoding uncharacterized XRE-cupin TFs were found near metabolism-related genes, illustrating the need of further systematic characterization of transcriptional regulatory networks in order to better understand the mechanisms of bacterial adaptation to new environments.IMPORTANCE Bacteria of the Pseudomonas genus, including the major human pathogen Pseudomonas aeruginosa, are known for their complex regulatory networks and high number of transcription factors, which contribute to their impressive adaptive ability. However, even in the most studied species, most of the regulators are still uncharacterized. With the recent advances in high-throughput sequencing methods, it is now possible to fill this knowledge gap and help the understanding of how bacteria adapt and thrive in new environments. By leveraging these methods, we provide an example of a comprehensive analysis of an entire family of transcription factors and bring new insights into metabolic and regulatory adaptation in the Pseudomonas genus.
Keywords: DAP-seq; Pseudomonas aeruginosa; RNA-seq; XRE-cupin; global regulatory networks; transcription factors.
Copyright © 2021 Trouillon et al.
Figures
Similar articles
-
Uncovering a hidden functional role of the XRE-cupin protein PsdR as a novel quorum-sensing regulator in Pseudomonas aeruginosa.PLoS Pathog. 2024 Mar 14;20(3):e1012078. doi: 10.1371/journal.ppat.1012078. eCollection 2024 Mar. PLoS Pathog. 2024. PMID: 38484003 Free PMC article.
-
A Novel XRE-Type Regulator Mediates Phage Lytic Development and Multiple Host Metabolic Processes in Pseudomonas aeruginosa.Microbiol Spectr. 2022 Dec 21;10(6):e0351122. doi: 10.1128/spectrum.03511-22. Epub 2022 Nov 29. Microbiol Spectr. 2022. PMID: 36445133 Free PMC article.
-
Determination of the two-component systems regulatory network reveals core and accessory regulations across Pseudomonas aeruginosa lineages.Nucleic Acids Res. 2021 Nov 18;49(20):11476-11490. doi: 10.1093/nar/gkab928. Nucleic Acids Res. 2021. PMID: 34718721 Free PMC article.
-
The transcriptional regulators of virulence for Pseudomonas aeruginosa: Therapeutic opportunity and preventive potential of its clinical infections.Genes Dis. 2022 Oct 1;10(5):2049-2063. doi: 10.1016/j.gendis.2022.09.009. eCollection 2023 Sep. Genes Dis. 2022. PMID: 37492705 Free PMC article. Review.
-
Virulence-related regulatory network of Pseudomonas syringae.Comput Struct Biotechnol J. 2022 Nov 9;20:6259-6270. doi: 10.1016/j.csbj.2022.11.011. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 36420163 Free PMC article. Review.
Cited by
-
An anticipatory mechanism enhances the cooperative behaviors of quorum sensing mutants in Pseudomonas aeruginosa.PLoS Pathog. 2025 Apr 15;21(4):e1013046. doi: 10.1371/journal.ppat.1013046. eCollection 2025 Apr. PLoS Pathog. 2025. PMID: 40233126 Free PMC article.
-
A brassinosteroid transcriptional regulatory network participates in regulating fiber elongation in cotton.Plant Physiol. 2023 Mar 17;191(3):1985-2000. doi: 10.1093/plphys/kiac590. Plant Physiol. 2023. PMID: 36542688 Free PMC article.
-
Predicting input signals of transcription factors in Escherichia coli.Mol Syst Biol. 2025 Jul 16. doi: 10.1038/s44320-025-00132-2. Online ahead of print. Mol Syst Biol. 2025. PMID: 40670652
-
The core and accessory Hfq interactomes across Pseudomonas aeruginosa lineages.Nat Commun. 2022 Mar 10;13(1):1258. doi: 10.1038/s41467-022-28849-w. Nat Commun. 2022. PMID: 35273147 Free PMC article.
-
Uncovering a hidden functional role of the XRE-cupin protein PsdR as a novel quorum-sensing regulator in Pseudomonas aeruginosa.PLoS Pathog. 2024 Mar 14;20(3):e1012078. doi: 10.1371/journal.ppat.1012078. eCollection 2024 Mar. PLoS Pathog. 2024. PMID: 38484003 Free PMC article.
References
-
- Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964. doi:10.1038/35023079. - DOI - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
