

FADU: a Quantification Tool for Prokaryotic Transcriptomic Analyses
- PMID: 33436511
- PMCID: PMC7901478
- DOI: 10.1128/mSystems.00917-20
FADU: a Quantification Tool for Prokaryotic Transcriptomic Analyses
Abstract
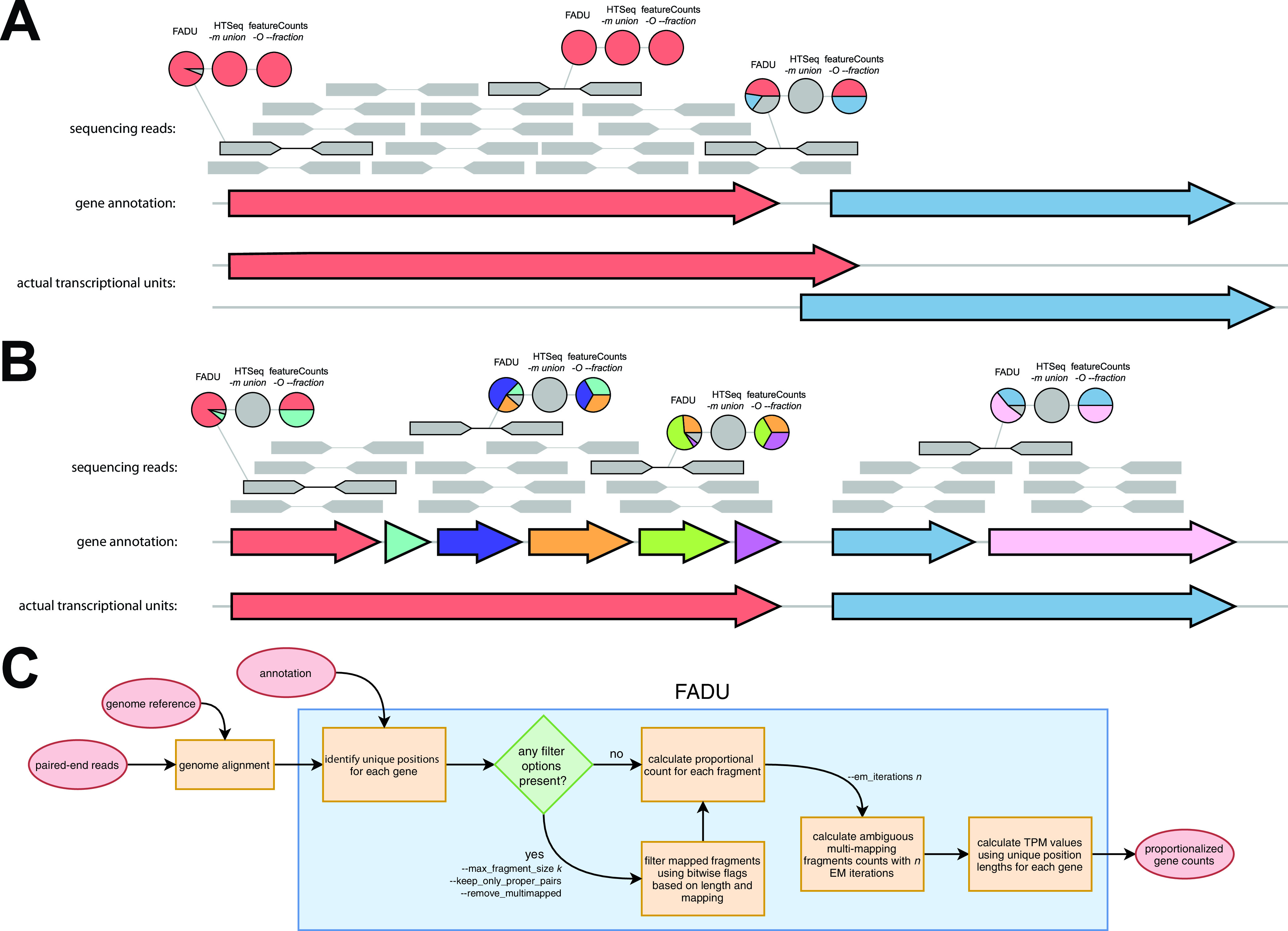
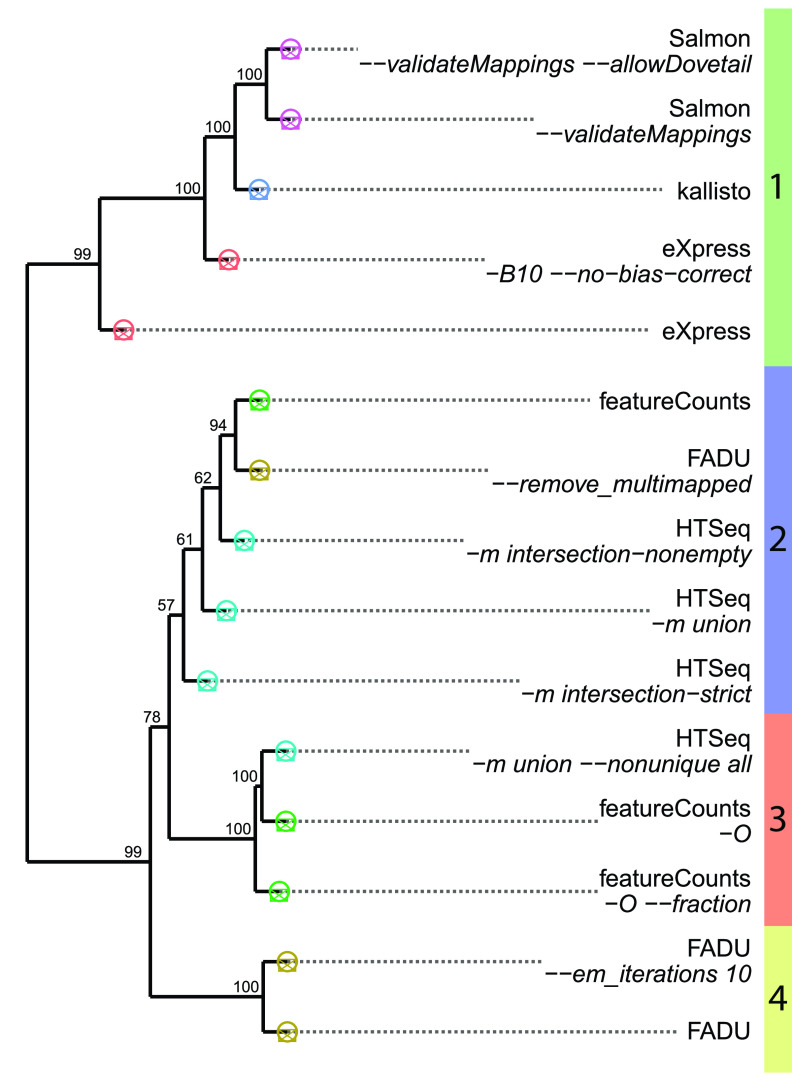
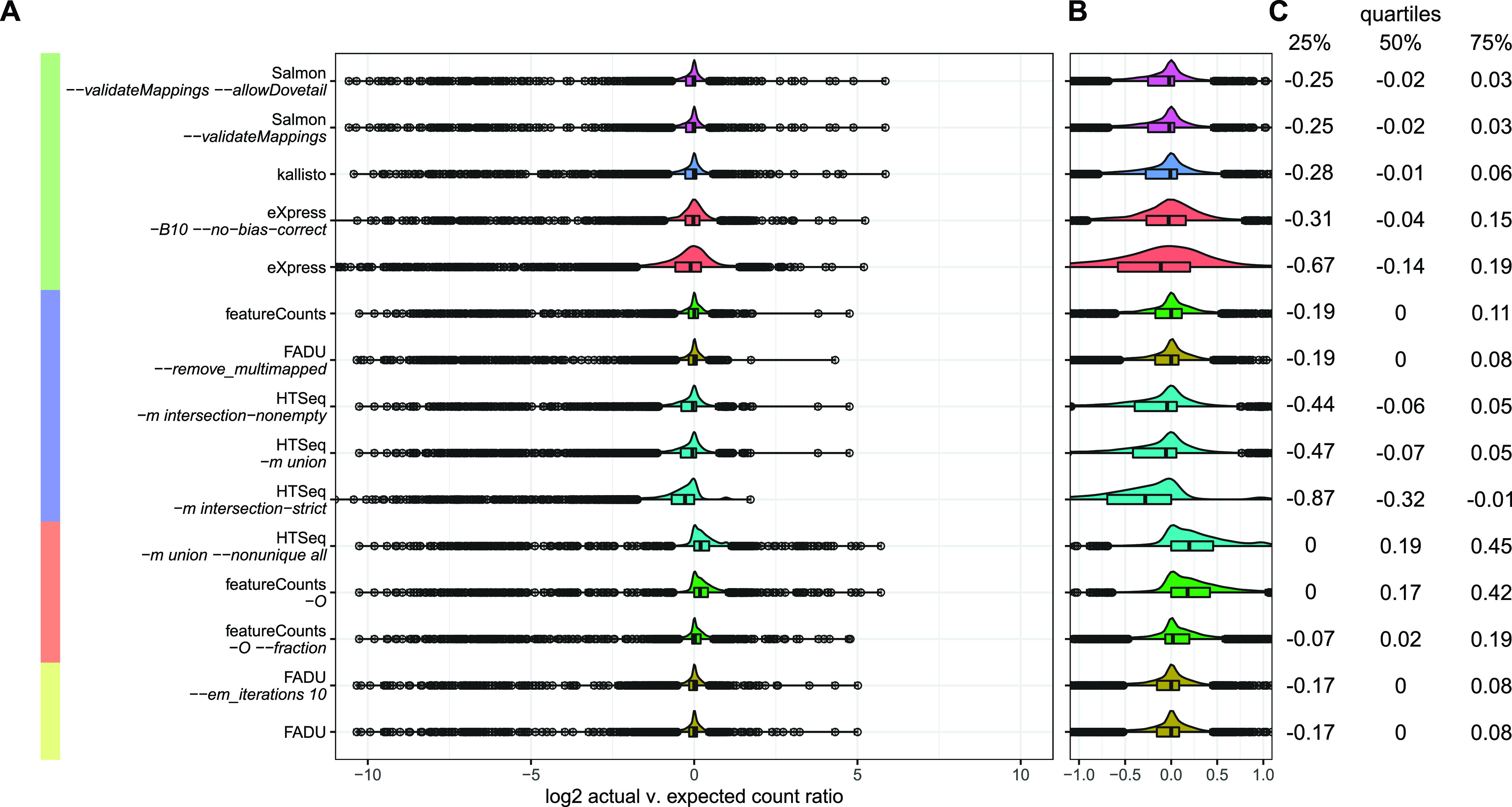
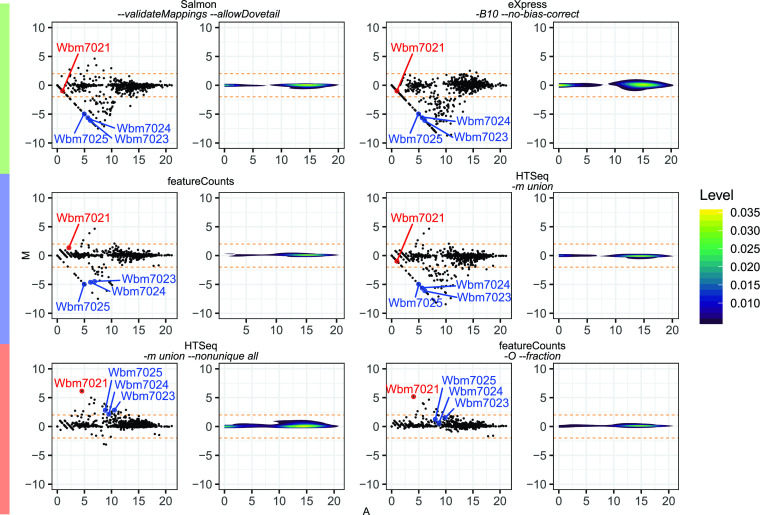
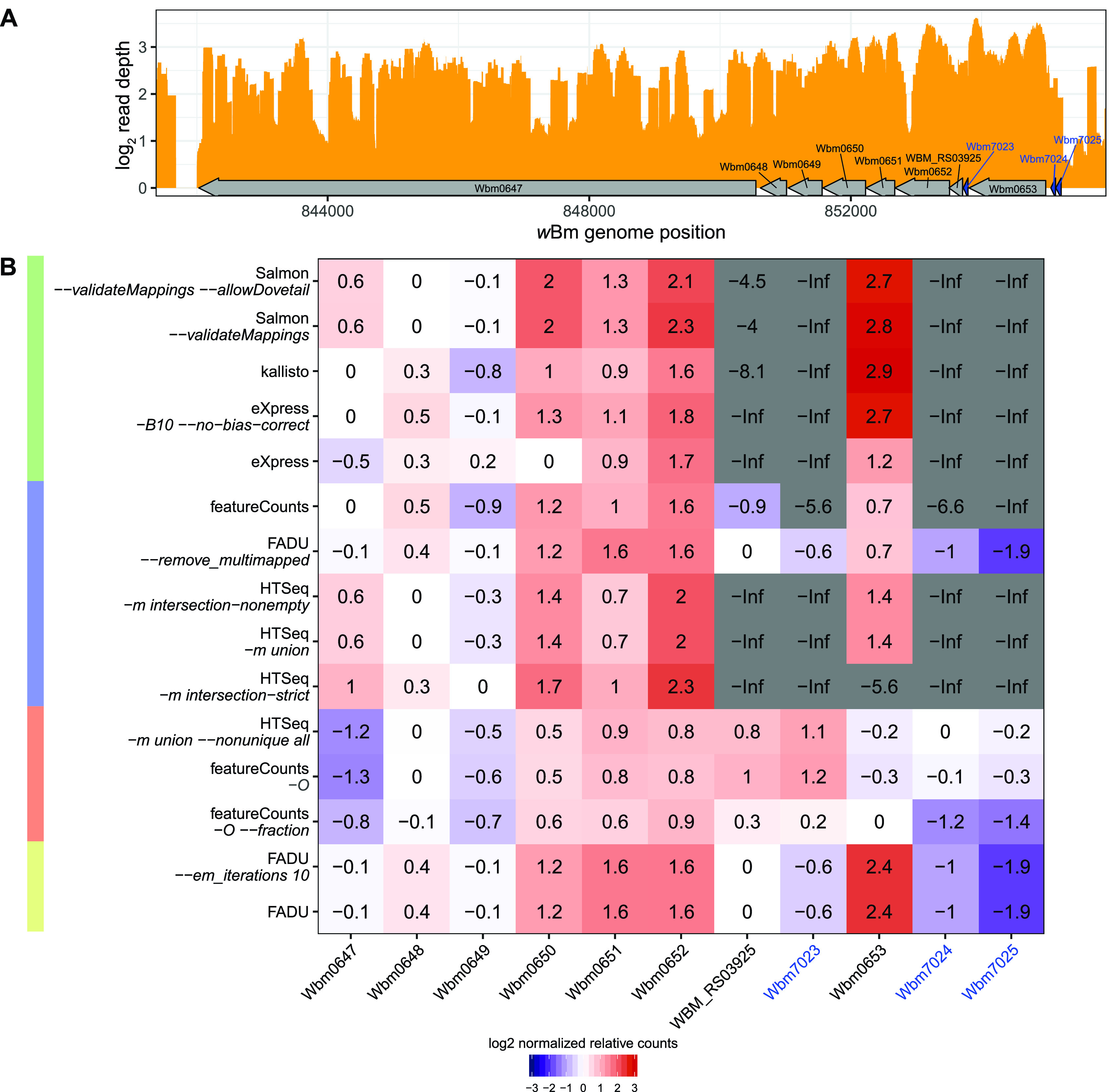
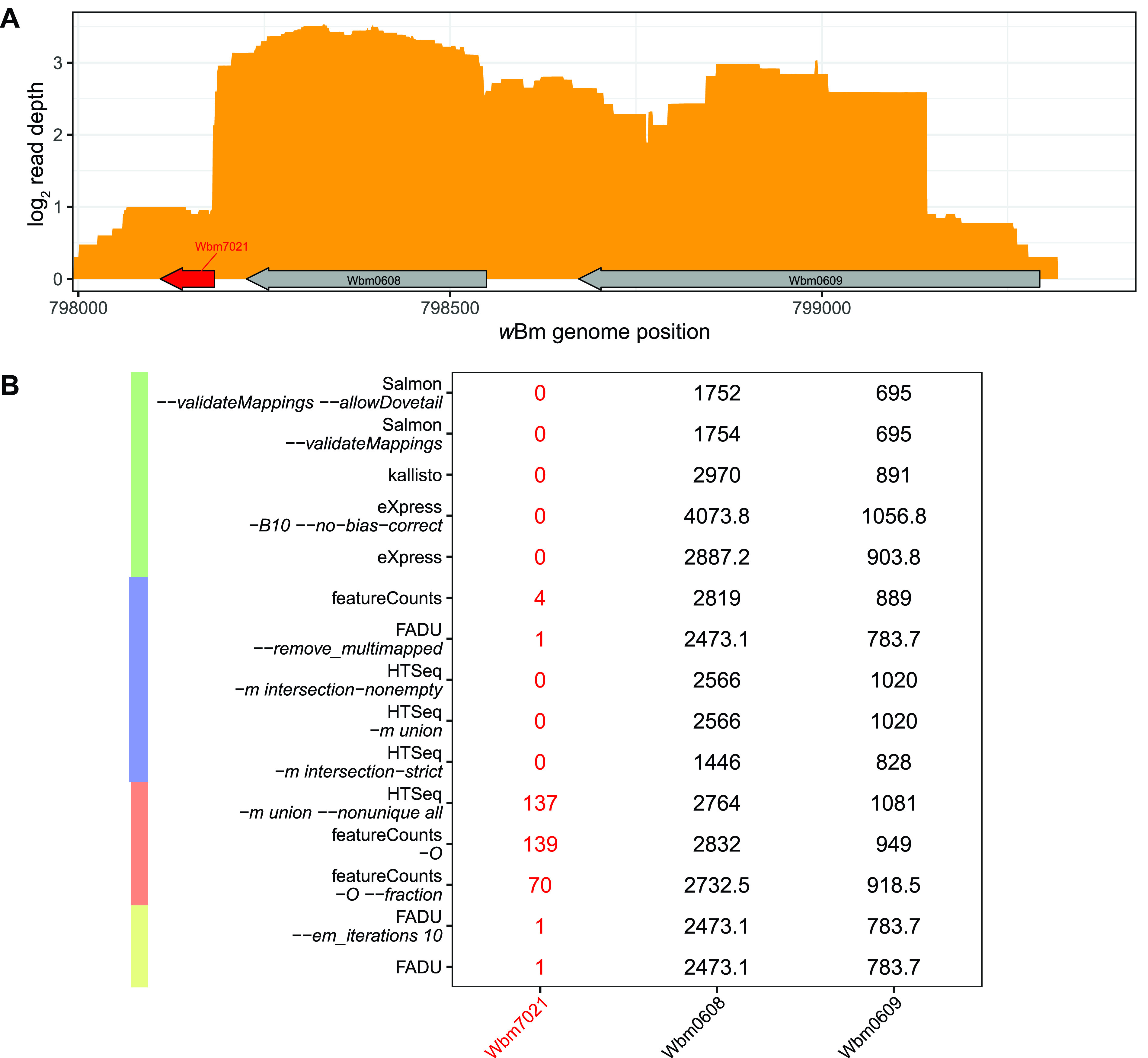
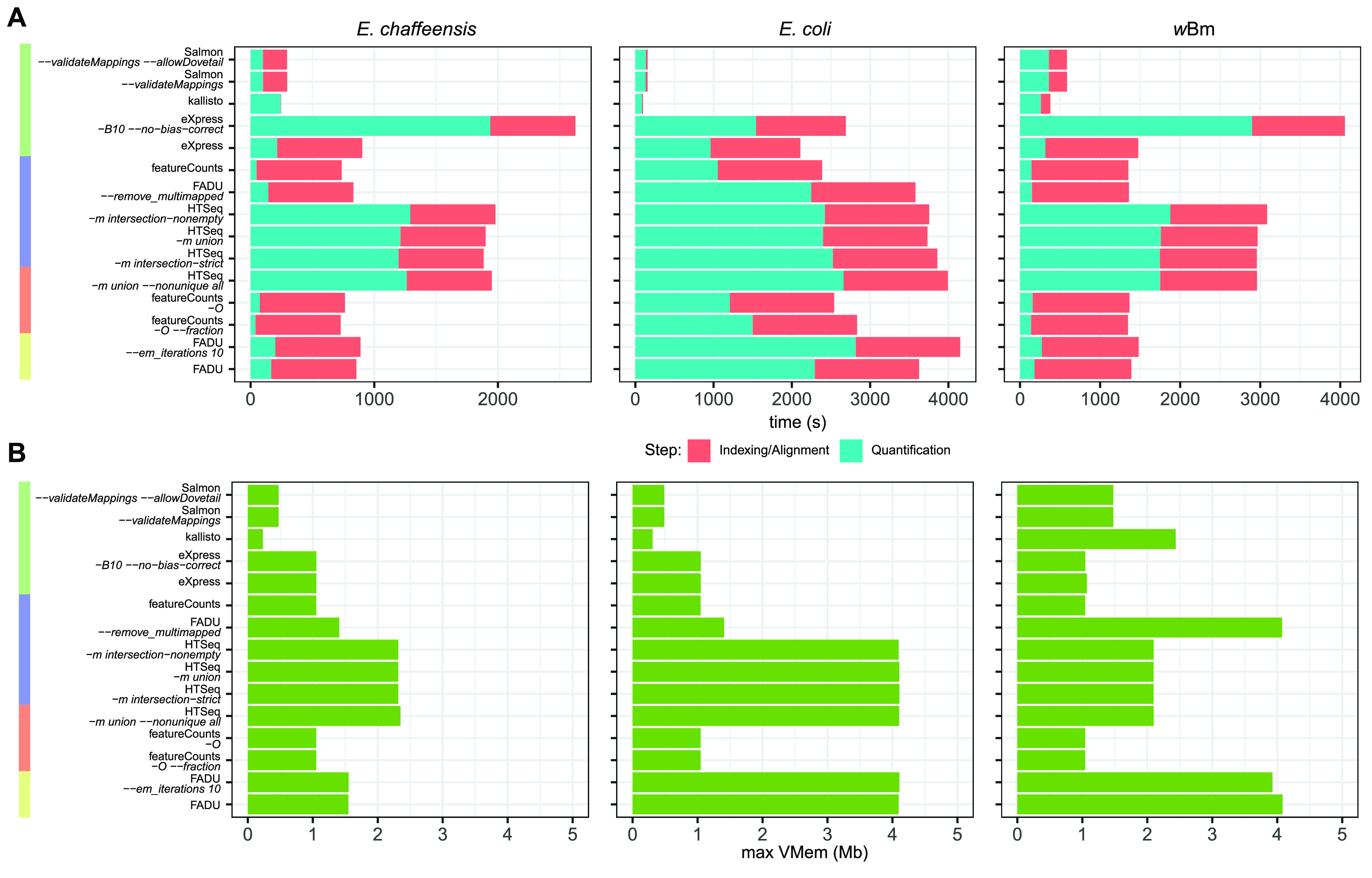
Quantification tools for RNA sequencing (RNA-Seq) analyses are often designed and tested using human transcriptomics data sets, in which full-length transcript sequences are well annotated. For prokaryotic transcriptomics experiments, full-length transcript sequences are seldom known, and coding sequences must instead be used for quantification steps in RNA-Seq analyses. However, operons confound accurate quantification of coding sequences since a single transcript does not necessarily equate to a single gene. Here, we introduce FADU (Feature Aggregate Depth Utility), a quantification tool designed specifically for prokaryotic RNA-Seq analyses. FADU assigns partial count values proportional to the length of the fragment overlapping the target feature. To assess the ability of FADU to quantify genes in prokaryotic transcriptomics analyses, we compared its performance to those of eXpress, featureCounts, HTSeq, kallisto, and Salmon across three paired-end read data sets of (i) Ehrlichia chaffeensis, (ii) Escherichia coli, and (iii) the Wolbachia endosymbiont wBm. Across each of the three data sets, we find that FADU can more accurately quantify operonic genes by deriving proportional counts for multigene fragments within operons. FADU is available at https://github.com/IGS/FADUIMPORTANCE Most currently available quantification tools for transcriptomics analyses have been designed for human data sets, in which full-length transcript sequences, including the untranslated regions, are well annotated. In most prokaryotic systems, full-length transcript sequences have yet to be characterized, leading to prokaryotic transcriptomics analyses being performed based on only the coding sequences. In contrast to eukaryotes, prokaryotes contain polycistronic transcripts, and when genes are quantified based on coding sequences instead of transcript sequences, this leads to an increased abundance of improperly assigned ambiguous multigene fragments, specifically those mapping to multiple genes in operons. Here, we describe FADU, a quantification tool for prokaryotic RNA-Seq analyses designed to assign proportional counts with the purpose of better quantifying operonic genes while minimizing the pitfalls associated with improperly assigning fragment counts from ambiguous transcripts.
Keywords: bacteria; differential expression; operon; polycistronic transcripts; read count; software; transcriptome; transcriptomics.
Copyright © 2021 Chung et al.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
