

Mechanisms of muscle atrophy and hypertrophy: implications in health and disease
- PMID: 33436614
- PMCID: PMC7803748
- DOI: 10.1038/s41467-020-20123-1
Mechanisms of muscle atrophy and hypertrophy: implications in health and disease
Abstract
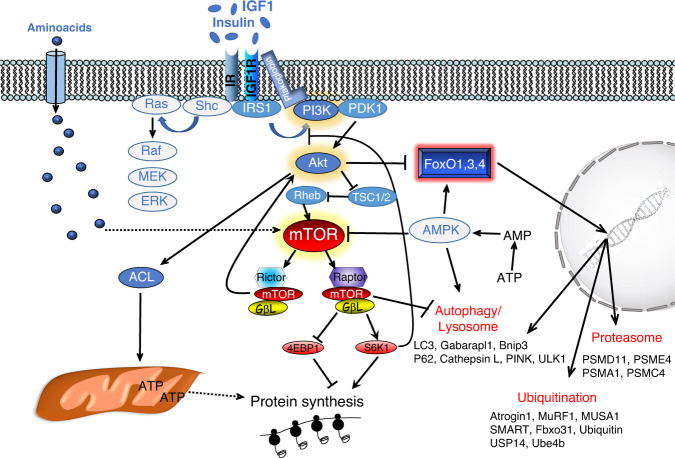
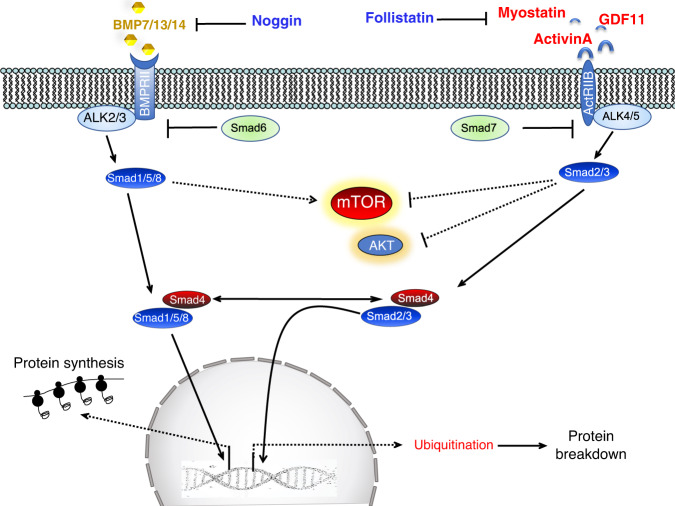
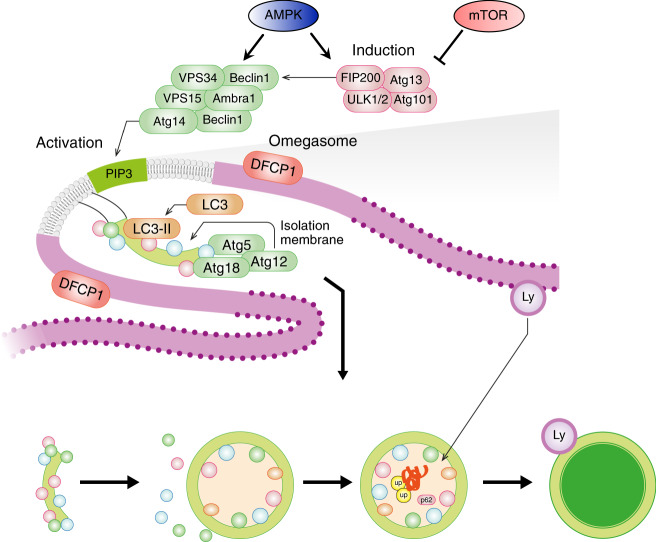
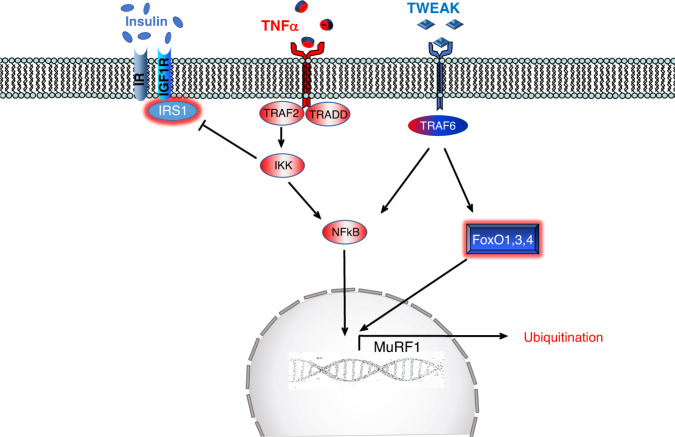
Skeletal muscle is the protein reservoir of our body and an important regulator of glucose and lipid homeostasis. Consequently, the growth or the loss of muscle mass can influence general metabolism, locomotion, eating and respiration. Therefore, it is not surprising that excessive muscle loss is a bad prognostic index of a variety of diseases ranging from cancer, organ failure, infections and unhealthy ageing. Muscle function is influenced by different quality systems that regulate the function of contractile proteins and organelles. These systems are controlled by transcriptional dependent programs that adapt muscle cells to environmental and nutritional clues. Mechanical, oxidative, nutritional and energy stresses, as well as growth factors or cytokines modulate signaling pathways that, ultimately, converge on protein and organelle turnover. Novel insights that control and orchestrate such complex network are continuously emerging and will be summarized in this review. Understanding the mechanisms that control muscle mass will provide therapeutic targets for the treatment of muscle loss in inherited and non-hereditary diseases and for the improvement of the quality of life during ageing.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
