

Characterizing genetic and antigenic divergence from vaccine strain of influenza A and B viruses circulating in Thailand, 2017-2020
- PMID: 33437008
- PMCID: PMC7803983
- DOI: 10.1038/s41598-020-80895-w
Characterizing genetic and antigenic divergence from vaccine strain of influenza A and B viruses circulating in Thailand, 2017-2020
Abstract
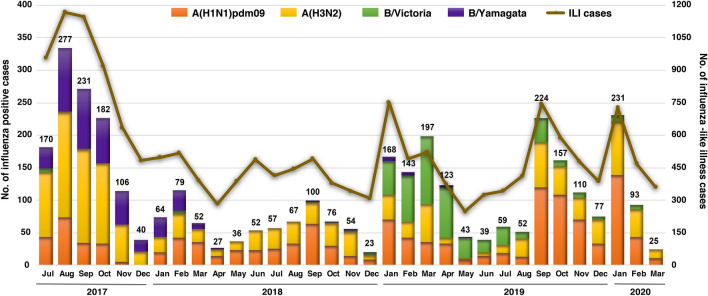
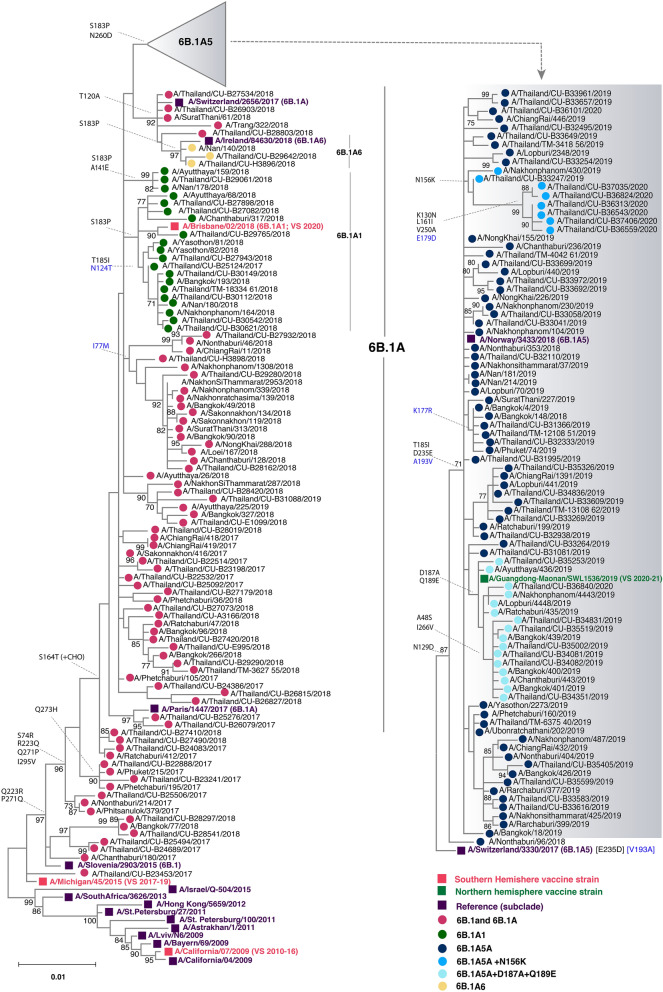
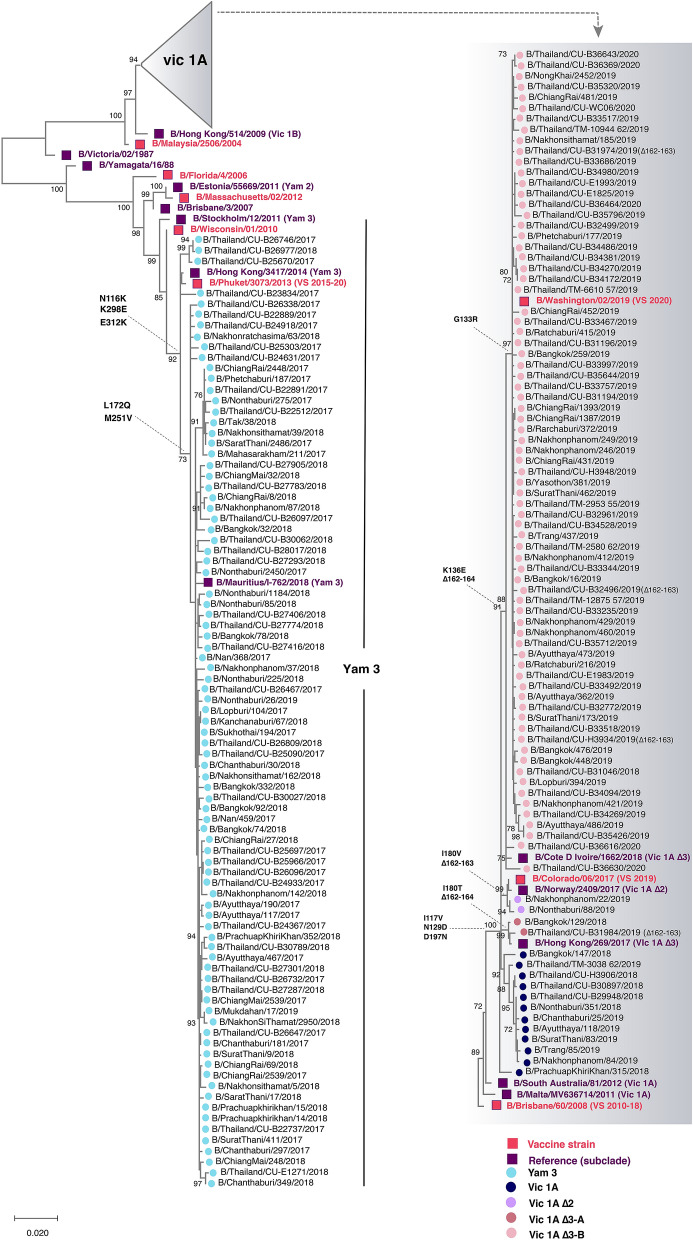
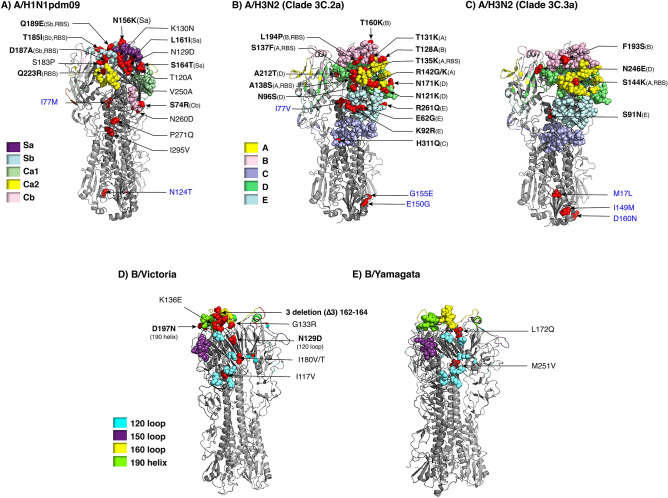
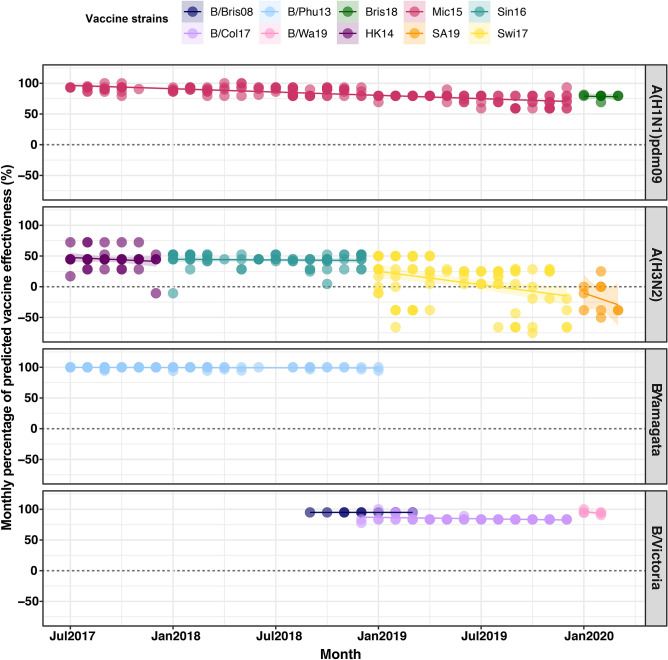
We monitored the circulating strains and genetic variation among seasonal influenza A and B viruses in Thailand between July 2017 and March 2020. The hemagglutinin gene was amplified and sequenced. We identified amino acid (AA) changes and computed antigenic relatedness using the Pepitope model. Phylogenetic analyses revealed multiple clades/subclades of influenza A(H1N1)pdm09 and A(H3N2) were circulating simultaneously and evolved away from their vaccine strain, but not the influenza B virus. The predominant circulating strains of A(H1N1)pdm09 belonged to 6B.1A1 (2017-2018) and 6B.1A5 (2019-2020) with additional AA substitutions. Clade 3C.2a1b and 3C.2a2 viruses co-circulated in A(H3N2) and clade 3C.3a virus was found in 2020. The B/Victoria-like lineage predominated since 2019 with an additional three AA deletions. Antigenic drift was dominantly facilitated at epitopes Sa and Sb of A(H1N1)pdm09, epitopes A, B, D and E of A(H3N2), and the 120 loop and 190 helix of influenza B virus. Moderate computed antigenic relatedness was observed in A(H1N1)pdm09. The computed antigenic relatedness of A(H3N2) indicated a significant decline in 2019 (9.17%) and 2020 (- 18.94%) whereas the circulating influenza B virus was antigenically similar (94.81%) with its vaccine strain. Our findings offer insights into the genetic divergence from vaccine strains, which could aid vaccine updating.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
