

Genomic diagnostics in polycystic kidney disease: an assessment of real-world use of whole-genome sequencing
- PMID: 33437033
- PMCID: PMC8110527
- DOI: 10.1038/s41431-020-00796-4
Genomic diagnostics in polycystic kidney disease: an assessment of real-world use of whole-genome sequencing
Abstract
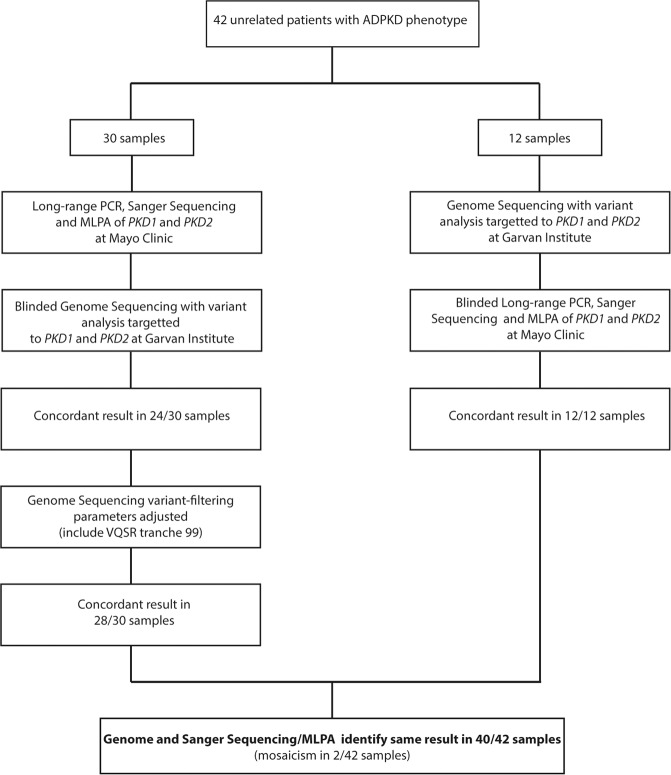
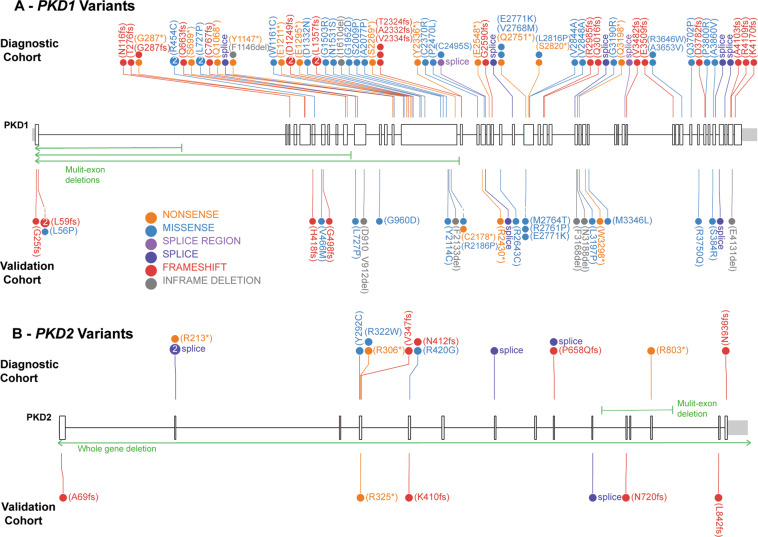
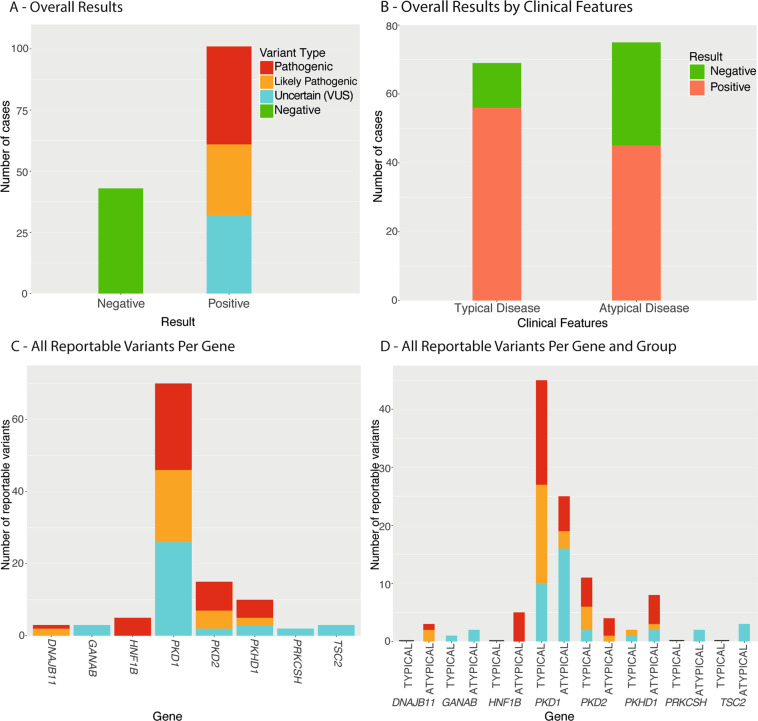
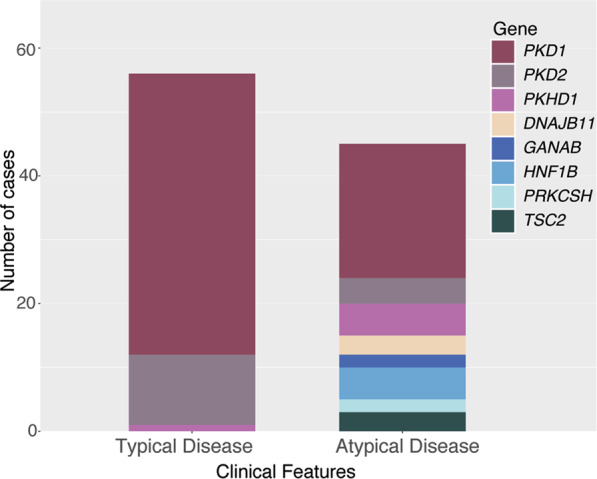
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is common, with a prevalence of 1/1000 and predominantly caused by disease-causing variants in PKD1 or PKD2. Clinical diagnosis is usually by age-dependent imaging criteria, which is challenging in patients with atypical clinical features, without family history, or younger age. However, there is increasing need for definitive diagnosis of ADPKD with new treatments available. Sequencing is complicated by six pseudogenes that share 97% homology to PKD1 and by recently identified phenocopy genes. Whole-genome sequencing can definitively diagnose ADPKD, but requires validation for clinical use. We initially performed a validation study, in which 42 ADPKD patients underwent sequencing of PKD1 and PKD2 by both whole-genome and Sanger sequencing, using a blinded, cross-over method. Whole-genome sequencing identified all PKD1 and PKD2 germline pathogenic variants in the validation study (sensitivity and specificity 100%). Two mosaic variants outside pipeline thresholds were not detected. We then examined the first 144 samples referred to a clinically-accredited diagnostic laboratory for clinical whole-genome sequencing, with targeted-analysis to a polycystic kidney disease gene-panel. In this unselected, diagnostic cohort (71 males :73 females), the diagnostic rate was 70%, including a diagnostic rate of 81% in patients with typical ADPKD (98% with PKD1/PKD2 variants) and 60% in those with atypical features (56% PKD1/PKD2; 44% PKHD1/HNF1B/GANAB/ DNAJB11/PRKCSH/TSC2). Most patients with atypical disease did not have clinical features that predicted likelihood of a genetic diagnosis. These results suggest clinicians should consider diagnostic genomics as part of their assessment in polycystic kidney disease, particularly in atypical disease.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
