

Streamlining data-intensive biology with workflow systems
- PMID: 33438730
- PMCID: PMC8631065
- DOI: 10.1093/gigascience/giaa140
Streamlining data-intensive biology with workflow systems
Abstract
As the scale of biological data generation has increased, the bottleneck of research has shifted from data generation to analysis. Researchers commonly need to build computational workflows that include multiple analytic tools and require incremental development as experimental insights demand tool and parameter modifications. These workflows can produce hundreds to thousands of intermediate files and results that must be integrated for biological insight. Data-centric workflow systems that internally manage computational resources, software, and conditional execution of analysis steps are reshaping the landscape of biological data analysis and empowering researchers to conduct reproducible analyses at scale. Adoption of these tools can facilitate and expedite robust data analysis, but knowledge of these techniques is still lacking. Here, we provide a series of strategies for leveraging workflow systems with structured project, data, and resource management to streamline large-scale biological analysis. We present these practices in the context of high-throughput sequencing data analysis, but the principles are broadly applicable to biologists working beyond this field.
Keywords: automation; data-intensive biology; repeatability; workflows.
© The Author(s) 2021. Published by Oxford University Press GigaScience.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
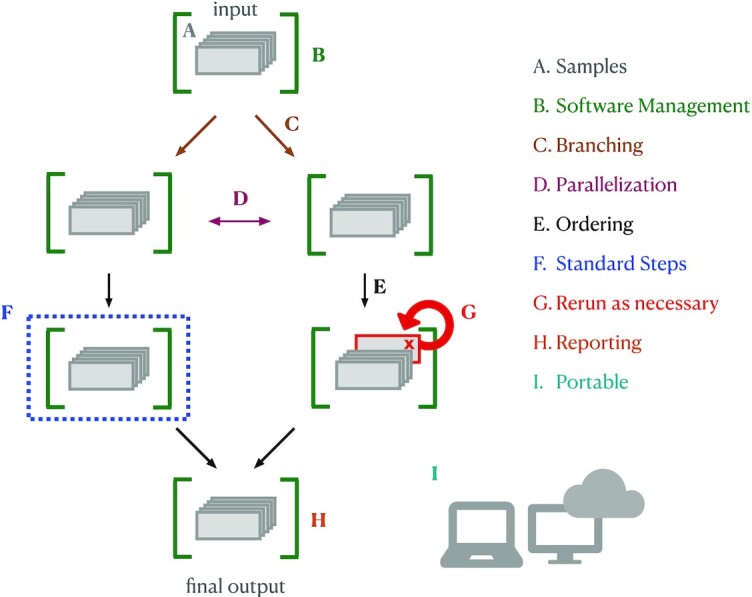
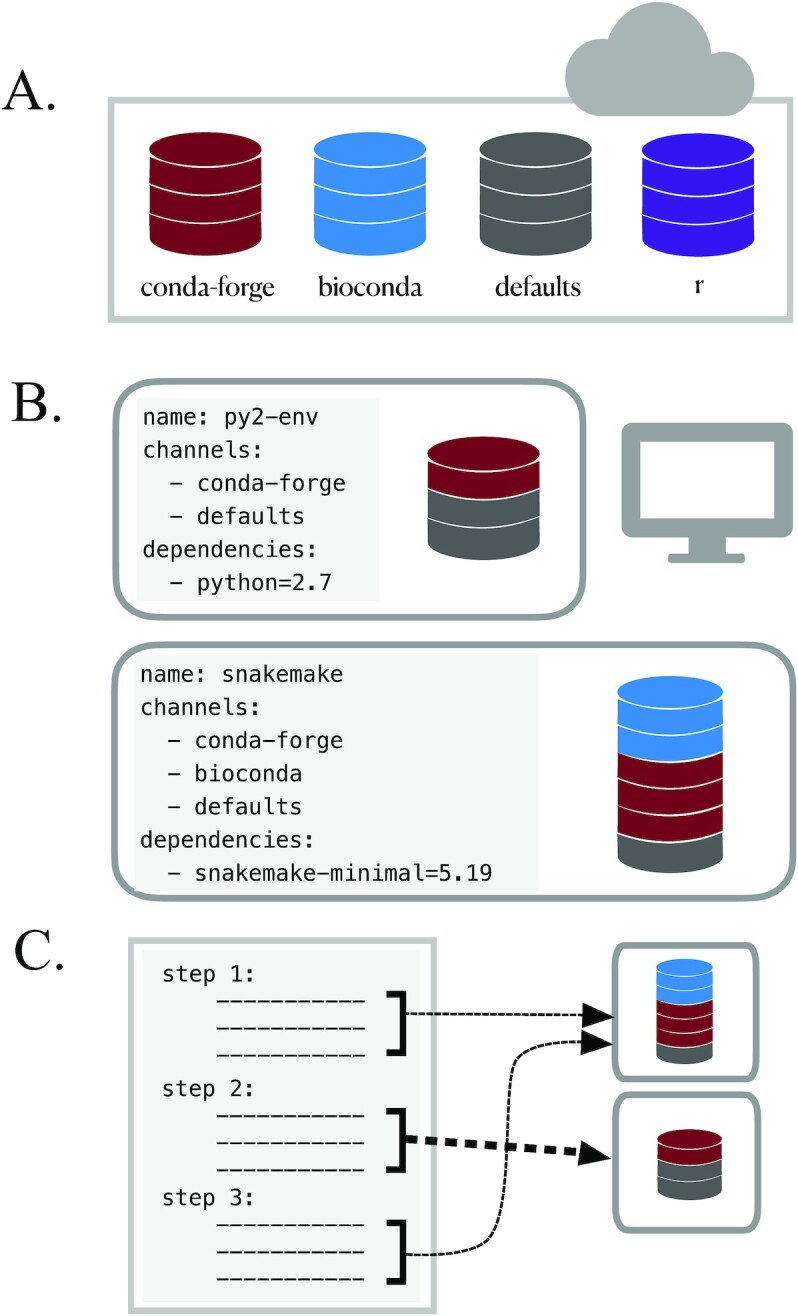
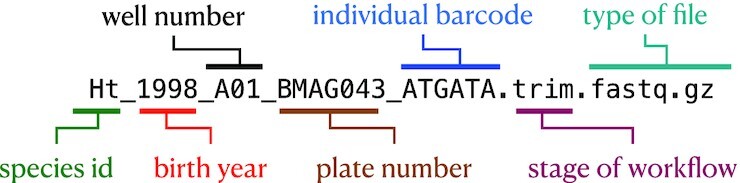
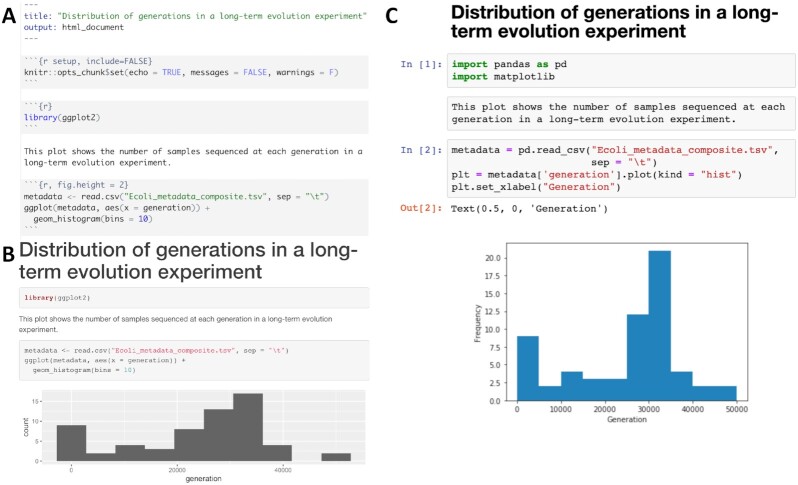
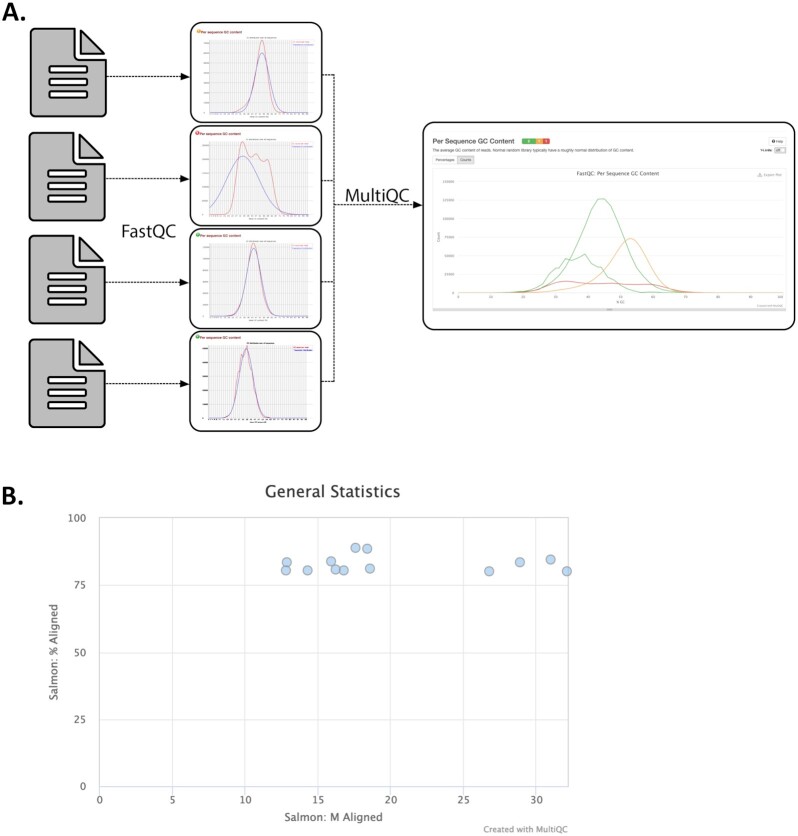
References
-
- Ewels PA, Peltzer A, Fillinger S, et al. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol. 2020;38(3):276–8. - PubMed
-
- Atkinson M, Gesing S, Montagnat J, et al. Scientific workflows: Past, present and future. Future Gener Comput Syst. 2017;75:216–27.
-
- Conery JS, Catchen JM, Lynch M. Rule-based workflow management for bioinformatics. VLDB J. 2005;14:318–29.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
