

RAD52 Adjusts Repair of Single-Strand Breaks via Reducing DNA-Damage-Promoted XRCC1/LIG3α Co-localization
- PMID: 33440161
- PMCID: PMC7872142
- DOI: 10.1016/j.celrep.2020.108625
RAD52 Adjusts Repair of Single-Strand Breaks via Reducing DNA-Damage-Promoted XRCC1/LIG3α Co-localization
Abstract
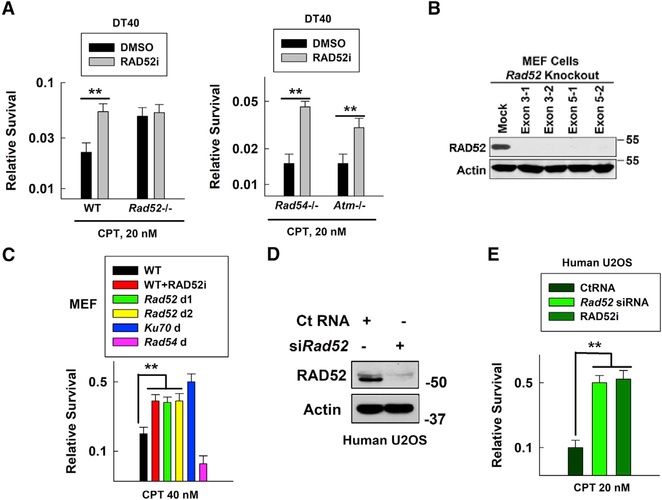
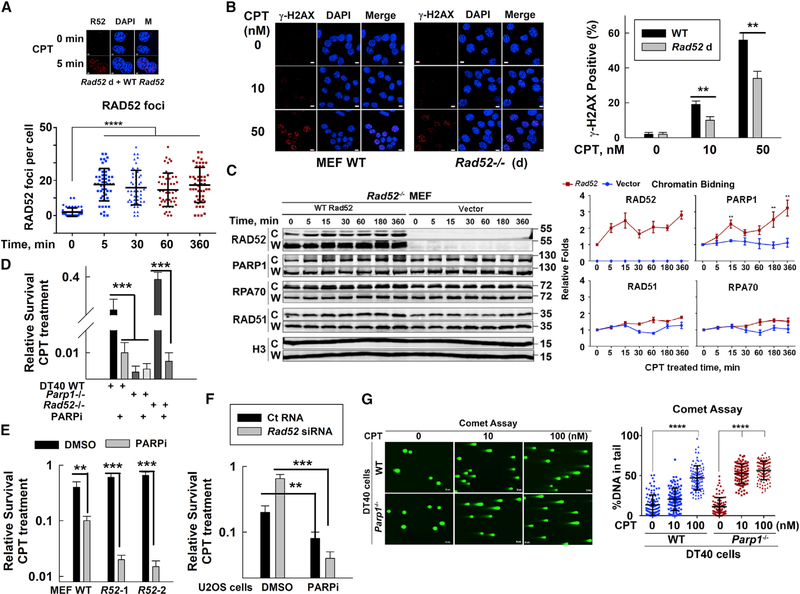
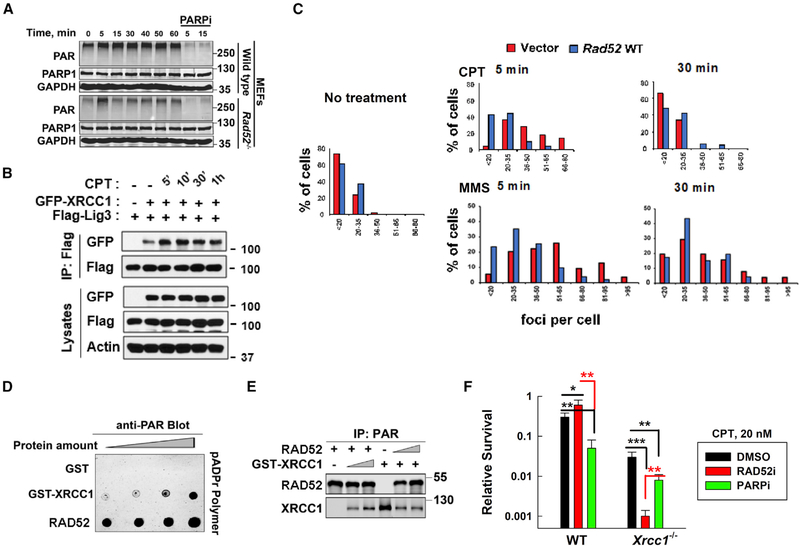
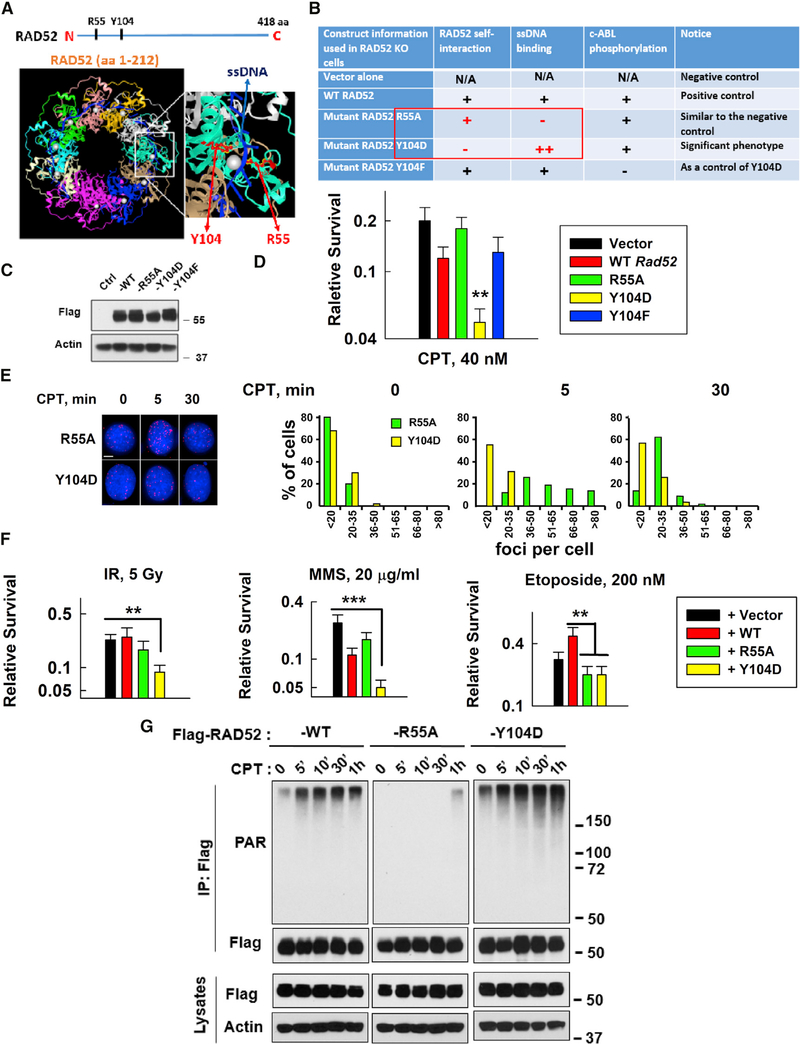
Radiation sensitive 52 (RAD52) is an important factor for double-strand break repair (DSBR). However, deficiency in vertebrate/mammalian Rad52 has no apparent phenotype. The underlying mechanism remains elusive. Here, we report that RAD52 deficiency increased cell survival after camptothecin (CPT) treatment. CPT generates single-strand breaks (SSBs) that further convert to double-strand breaks (DSBs) if they are not repaired. RAD52 inhibits SSB repair (SSBR) through strong single-strand DNA (ssDNA) and/or poly(ADP-ribose) (PAR) binding affinity to reduce DNA-damage-promoted X-Ray Repair Cross Complementing 1 (XRCC1)/ligase IIIα (LIG3α) co-localization. The inhibitory effects of RAD52 on SSBR neutralize the role of RAD52 in DSBR, suggesting that RAD52 may maintain a balance between cell survival and genomic integrity. Furthermore, we demonstrate that blocking RAD52 oligomerization that disrupts RAD52's DSBR, while retaining its ssDNA binding capacity that is required for RAD52's inhibitory effects on SSBR, sensitizes cells to different DNA-damaging agents. This discovery provides guidance for developing efficient RAD52 inhibitors in cancer therapy.
Keywords: DNA damage; DNA repair; RAD52; XRCC1; single-strand breaks.
Copyright © 2020 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
Similar articles
-
XRCC1 deficiency increased the DNA damage induced by γ-ray in HepG2 cell: Involvement of DSB repair and cell cycle arrest.Environ Toxicol Pharmacol. 2013 Sep;36(2):311-319. doi: 10.1016/j.etap.2013.04.009. Epub 2013 May 2. Environ Toxicol Pharmacol. 2013. PMID: 23708312
-
Replication Protein A Phosphorylation Facilitates RAD52-Dependent Homologous Recombination in BRCA-Deficient Cells.Mol Cell Biol. 2022 Feb 17;42(2):e0052421. doi: 10.1128/mcb.00524-21. Epub 2021 Dec 20. Mol Cell Biol. 2022. PMID: 34928169 Free PMC article.
-
Damage response of XRCC1 at sites of DNA single strand breaks is regulated by phosphorylation and ubiquitylation after degradation of poly(ADP-ribose).J Cell Sci. 2013 Oct 1;126(Pt 19):4414-23. doi: 10.1242/jcs.128272. Epub 2013 Jul 18. J Cell Sci. 2013. PMID: 23868975 Free PMC article.
-
XRCC1 protein; Form and function.DNA Repair (Amst). 2019 Sep;81:102664. doi: 10.1016/j.dnarep.2019.102664. Epub 2019 Jul 8. DNA Repair (Amst). 2019. PMID: 31324530 Review.
-
Mammalian DNA ligases; roles in maintaining genome integrity.J Mol Biol. 2024 Jan 1;436(1):168276. doi: 10.1016/j.jmb.2023.168276. Epub 2023 Sep 13. J Mol Biol. 2024. PMID: 37714297 Free PMC article. Review.
Cited by
-
ALDH1A1 drives prostate cancer metastases and radioresistance by interplay with AR- and RAR-dependent transcription.Theranostics. 2024 Jan 1;14(2):714-737. doi: 10.7150/thno.88057. eCollection 2024. Theranostics. 2024. PMID: 38169509 Free PMC article.
-
Identifying polymorphic cis-regulatory variants as risk markers for lung carcinogenesis and chemotherapy responses in tobacco smokers from eastern India.Sci Rep. 2023 Mar 10;13(1):4019. doi: 10.1038/s41598-023-30962-9. Sci Rep. 2023. PMID: 36899086 Free PMC article.
-
RAD52: Paradigm of Synthetic Lethality and New Developments.Front Genet. 2021 Nov 23;12:780293. doi: 10.3389/fgene.2021.780293. eCollection 2021. Front Genet. 2021. PMID: 34887904 Free PMC article. Review.
-
Genetic predictors of neurocognitive outcomes in survivors of pediatric brain tumors.J Neurooncol. 2023 Oct;165(1):161-169. doi: 10.1007/s11060-023-04472-7. Epub 2023 Oct 25. J Neurooncol. 2023. PMID: 37878192 Free PMC article.
-
The Influence of Circadian Rhythms on DNA Damage Repair in Skin Photoaging.Int J Mol Sci. 2024 Oct 11;25(20):10926. doi: 10.3390/ijms252010926. Int J Mol Sci. 2024. PMID: 39456709 Free PMC article. Review.
References
-
- Adachi N, So S, and Koyama H (2004). Loss of nonhomologous end joining confers camptothecin resistance in DT40 cells. Implications for the repair of topoisomerase I-mediated DNA damage. J. Biol. Chem 279, 37343–37348. - PubMed
-
- Boehler C, Gauthier L, Yelamos J, Noll A, Schreiber V, and Dantzer F (2011). Phenotypic characterization of Parp-1 and Parp-2 deficient mice and cells In Poly(ADP-ribose) Polymerase: Methods and Protocols, Tulin AV, ed. (Humana; ), pp. 313–336. - PubMed
-
- Caldecott KW (2008). Single-strand break repair and genetic disease. Nat. Rev. Genet. 9, 619–631. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
