

Disentangling selection on genetically correlated polygenic traits via whole-genome genealogies
- PMID: 33440170
- PMCID: PMC7895848
- DOI: 10.1016/j.ajhg.2020.12.005
Disentangling selection on genetically correlated polygenic traits via whole-genome genealogies
Abstract
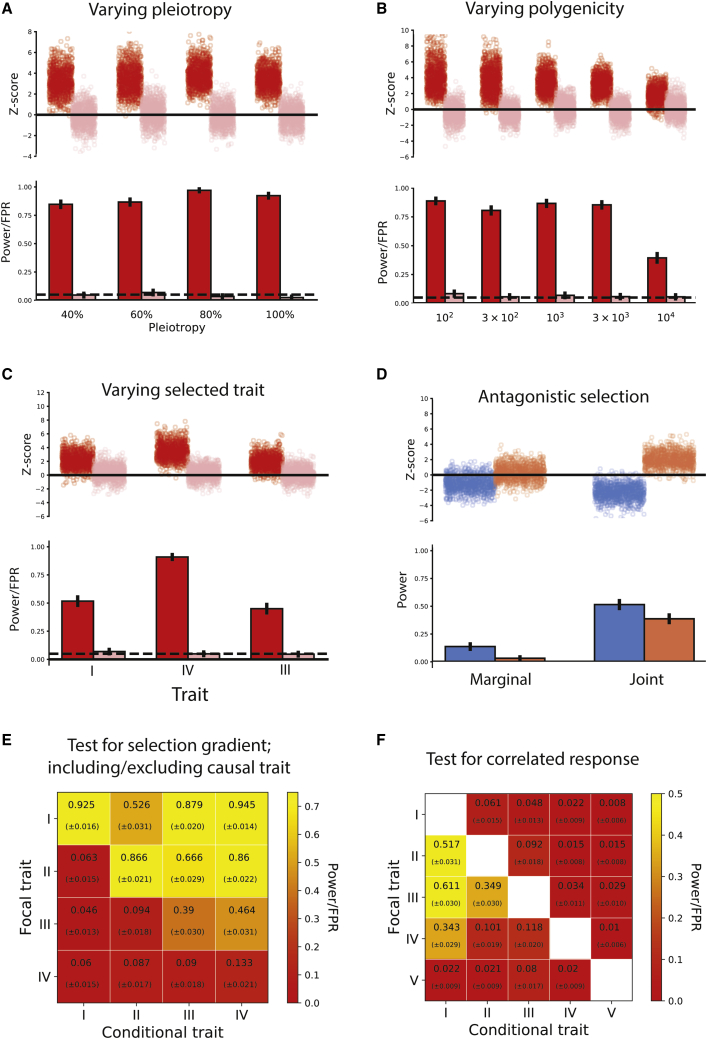
We present a full-likelihood method to infer polygenic adaptation from DNA sequence variation and GWAS summary statistics to quantify recent transient directional selection acting on a complex trait. Through simulations of polygenic trait architecture evolution and GWASs, we show the method substantially improves power over current methods. We examine the robustness of the method under stratification, uncertainty and bias in marginal effects, uncertainty in the causal SNPs, allelic heterogeneity, negative selection, and low GWAS sample size. The method can quantify selection acting on correlated traits, controlling for pleiotropy even among traits with strong genetic correlation (|rg|=80%) while retaining high power to attribute selection to the causal trait. When the causal trait is excluded from analysis, selection is attributed to its closest proxy. We discuss limitations of the method, cautioning against strongly causal interpretations of the results, and the possibility of undetectable gene-by-environment (GxE) interactions. We apply the method to 56 human polygenic traits, revealing signals of directional selection on pigmentation, life history, glycated hemoglobin (HbA1c), and other traits. We also conduct joint testing of 137 pairs of genetically correlated traits, revealing widespread correlated response acting on these traits (2.6-fold enrichment, p = 1.5 × 10-7). Signs of selection on some traits previously reported as adaptive (e.g., educational attainment and hair color) are largely attributable to correlated response (p = 2.9 × 10-6 and 1.7 × 10-4, respectively). Lastly, our joint test shows antagonistic selection has increased type 2 diabetes risk and decrease HbA1c (p = 1.5 × 10-5).
Keywords: GWAS; adaptation; ancestral recombination graph; polygenic selection; stratification; thrifty gene.
Copyright © 2020 American Society of Human Genetics. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Loh P.-R., Bhatia G., Gusev A., Finucane H.K., Bulik-Sullivan B.K., Pollack S.J., de Candia T.R., Lee S.H., Wray N.R., Kendler K.S., Schizophrenia Working Group of Psychiatric Genomics Consortium Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat. Genet. 2015;47:1385–1392. - PMC - PubMed
-
- Zeng J., de Vlaming R., Wu Y., Robinson M.R., Lloyd-Jones L.R., Yengo L., Yap C.X., Xue A., Sidorenko J., McRae A.F. Signatures of negative selection in the genetic architecture of human complex traits. Nat. Genet. 2018;50:746–753. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
