

Patterns of de novo tandem repeat mutations and their role in autism
- PMID: 33442040
- PMCID: PMC7810352
- DOI: 10.1038/s41586-020-03078-7
Patterns of de novo tandem repeat mutations and their role in autism
Abstract
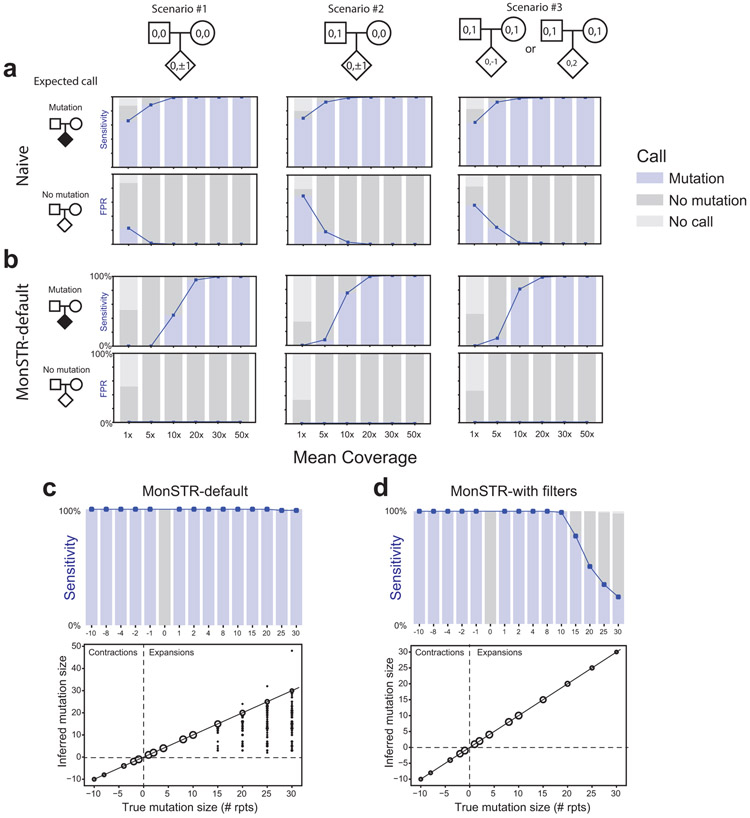
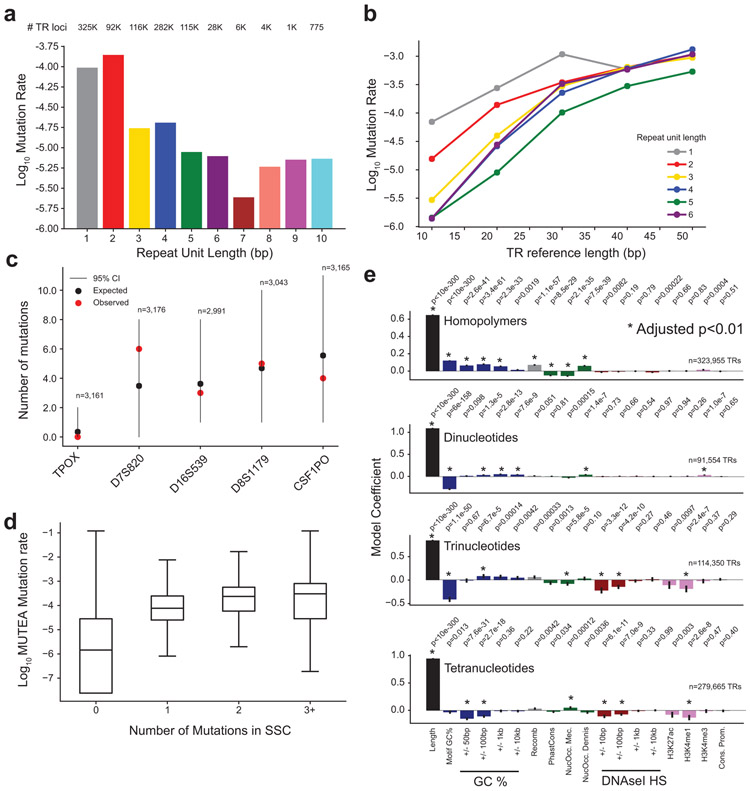
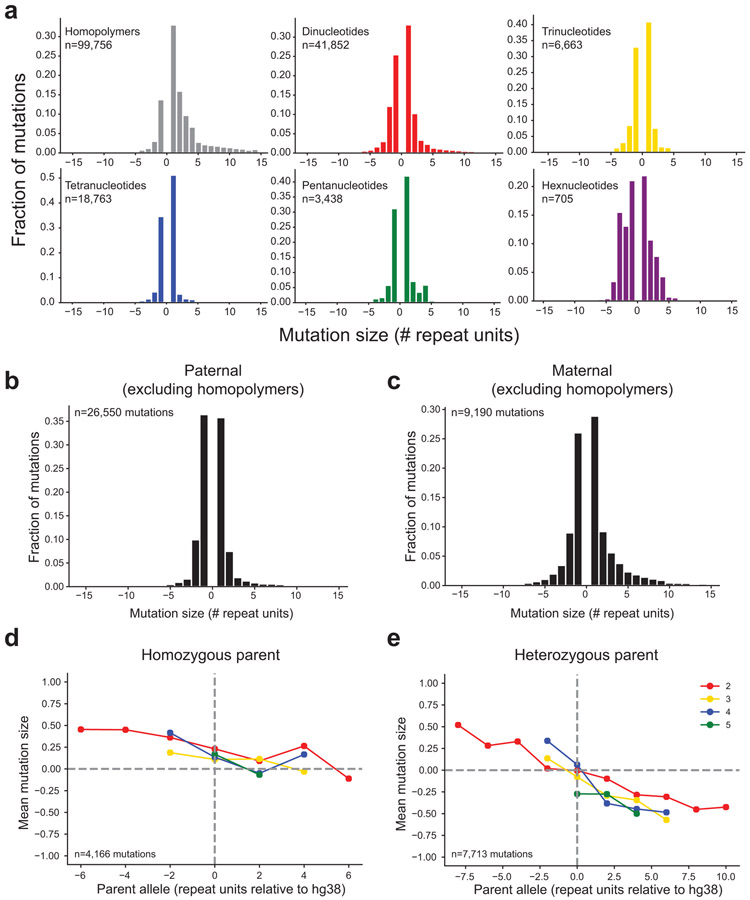
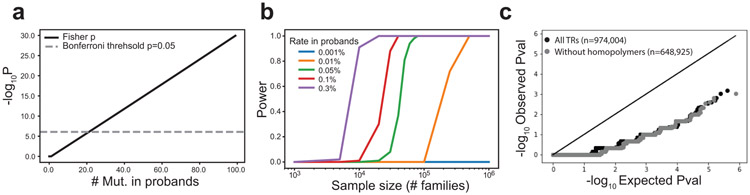
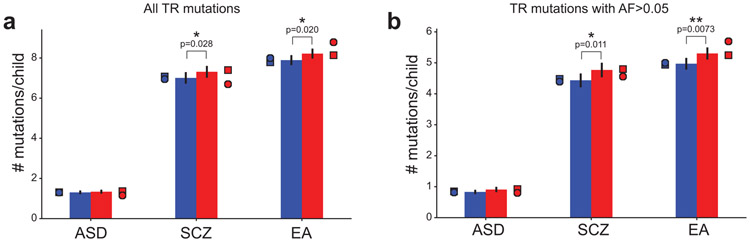
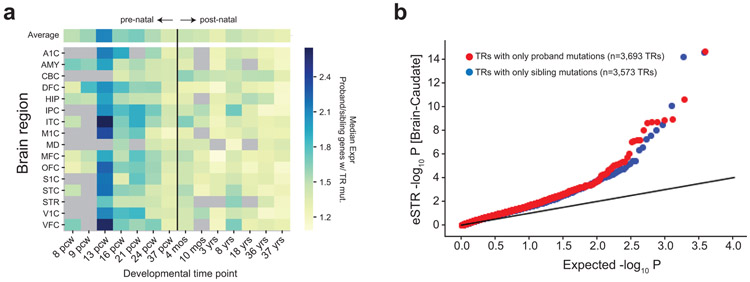
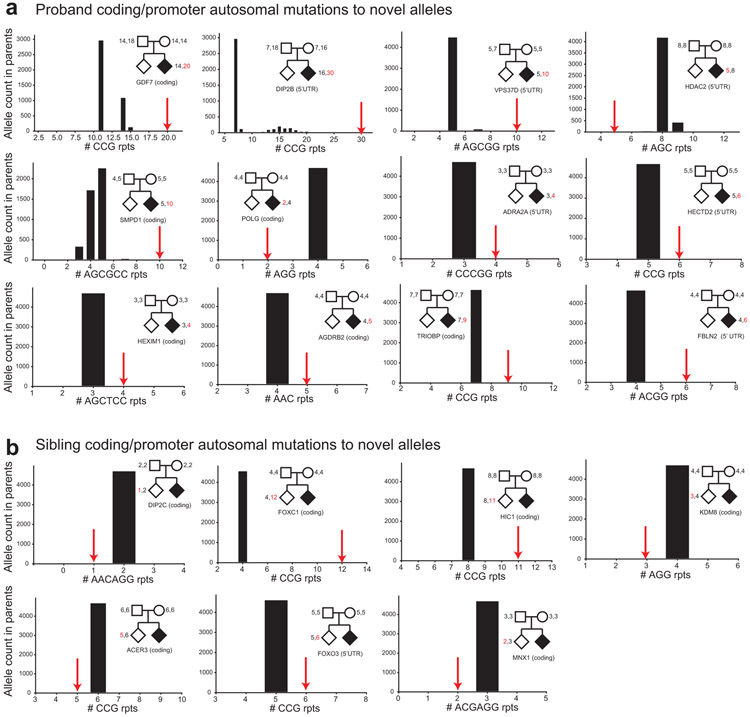
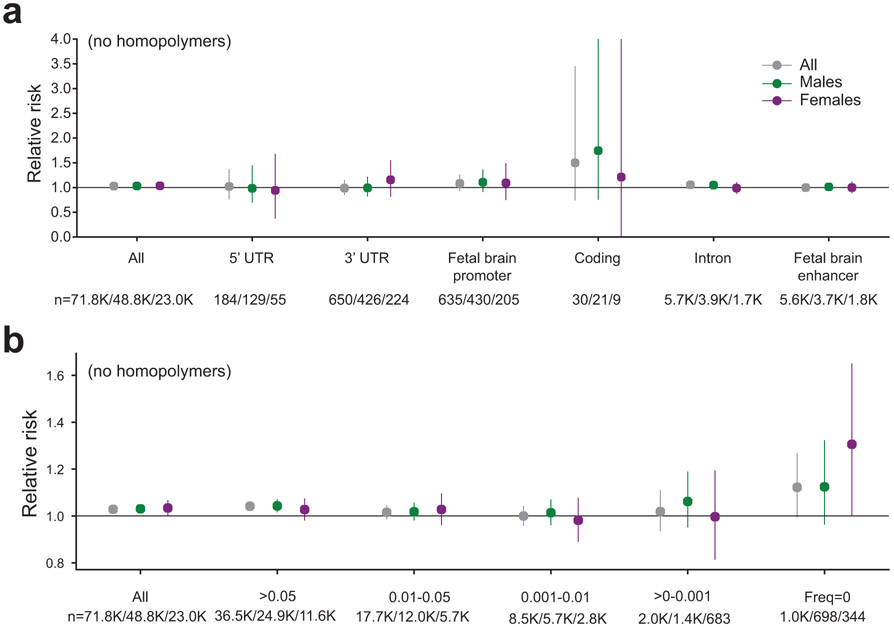
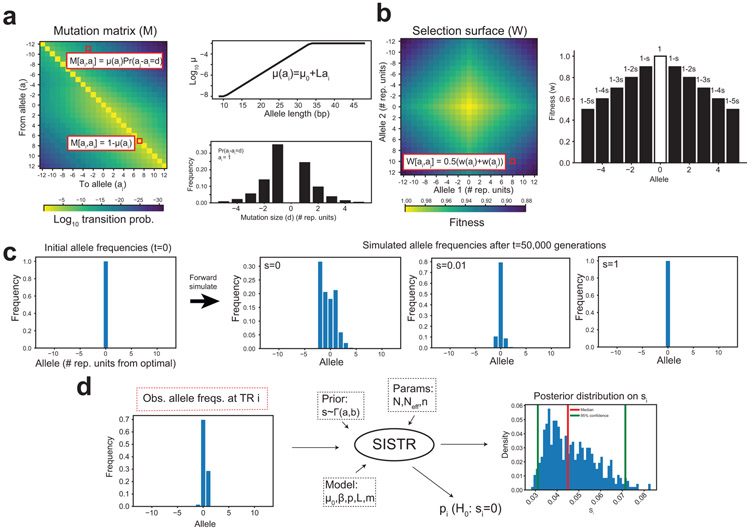
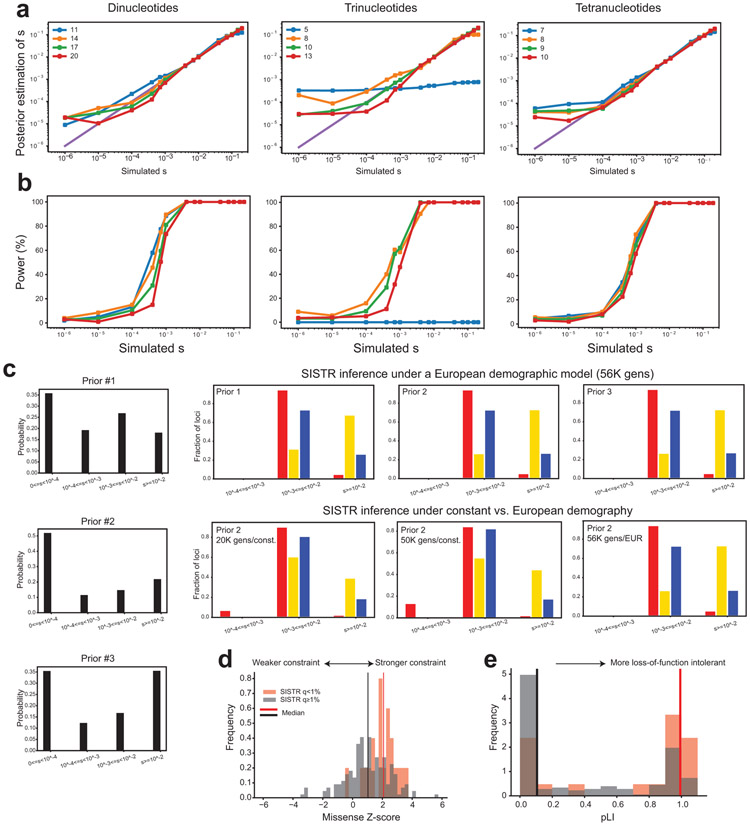
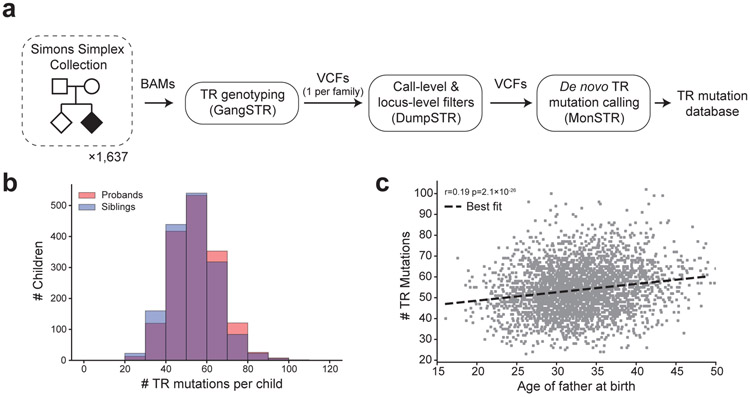
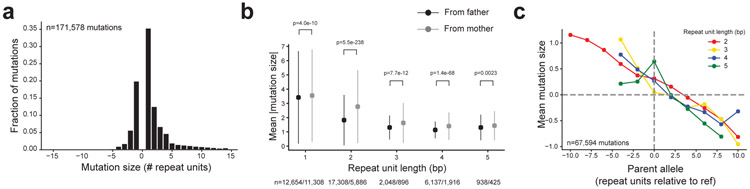
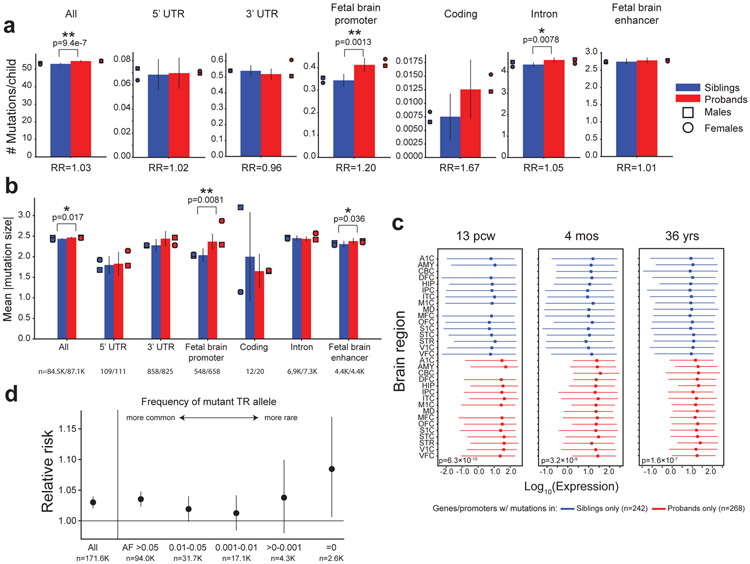
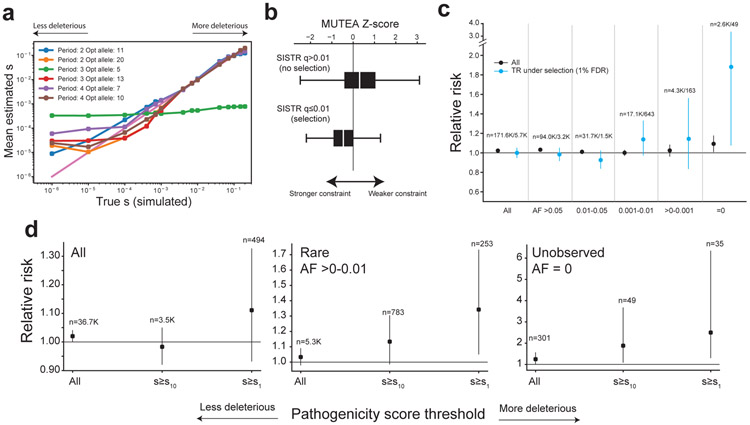
Autism spectrum disorder (ASD) is an early-onset developmental disorder characterized by deficits in communication and social interaction and restrictive or repetitive behaviours1,2. Family studies demonstrate that ASD has a substantial genetic basis with contributions both from inherited and de novo variants3,4. It has been estimated that de novo mutations may contribute to 30% of all simplex cases, in which only a single child is affected per family5. Tandem repeats (TRs), defined here as sequences of 1 to 20 base pairs in size repeated consecutively, comprise one of the major sources of de novo mutations in humans6. TR expansions are implicated in dozens of neurological and psychiatric disorders7. Yet, de novo TR mutations have not been characterized on a genome-wide scale, and their contribution to ASD remains unexplored. Here we develop new bioinformatics methods for identifying and prioritizing de novo TR mutations from sequencing data and perform a genome-wide characterization of de novo TR mutations in ASD-affected probands and unaffected siblings. We infer specific mutation events and their precise changes in repeat number, and primarily focus on more prevalent stepwise copy number changes rather than large expansions. Our results demonstrate a significant genome-wide excess of TR mutations in ASD probands. Mutations in probands tend to be larger, enriched in fetal brain regulatory regions, and are predicted to be more evolutionarily deleterious. Overall, our results highlight the importance of considering repeat variants in future studies of de novo mutations.
Figures
Comment in
-
Repeat DNA expands our understanding of autism spectrum disorder.Nature. 2021 Jan;589(7841):200-202. doi: 10.1038/d41586-020-03658-7. Nature. 2021. PMID: 33442037 No abstract available.
-
Linking newly occurring mutations to autism.Nat Rev Genet. 2021 Mar;22(3):133. doi: 10.1038/s41576-021-00335-x. Nat Rev Genet. 2021. PMID: 33542502 No abstract available.
References
Main References
-
- Association, A. P. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®). (American Psychiatric Pub, 2013).
Methods References
-
- Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv: Genomics (2013).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
