

The Parkinson's disease-associated gene ITPKB protects against α-synuclein aggregation by regulating ER-to-mitochondria calcium release
- PMID: 33443159
- PMCID: PMC7817155
- DOI: 10.1073/pnas.2006476118
The Parkinson's disease-associated gene ITPKB protects against α-synuclein aggregation by regulating ER-to-mitochondria calcium release
Abstract
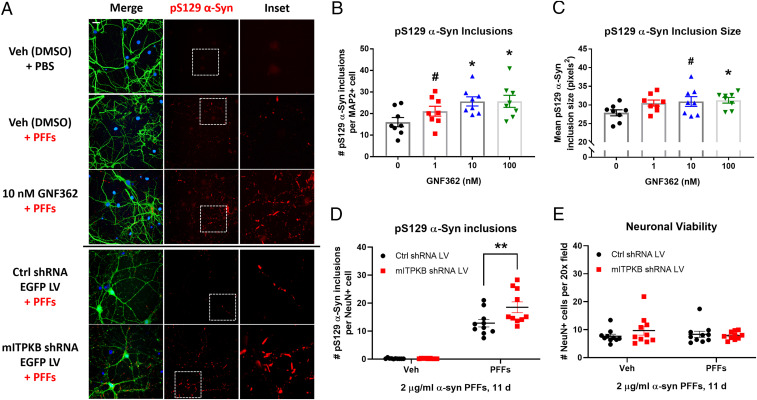
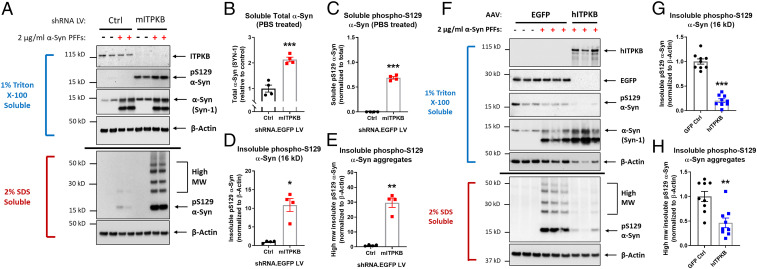
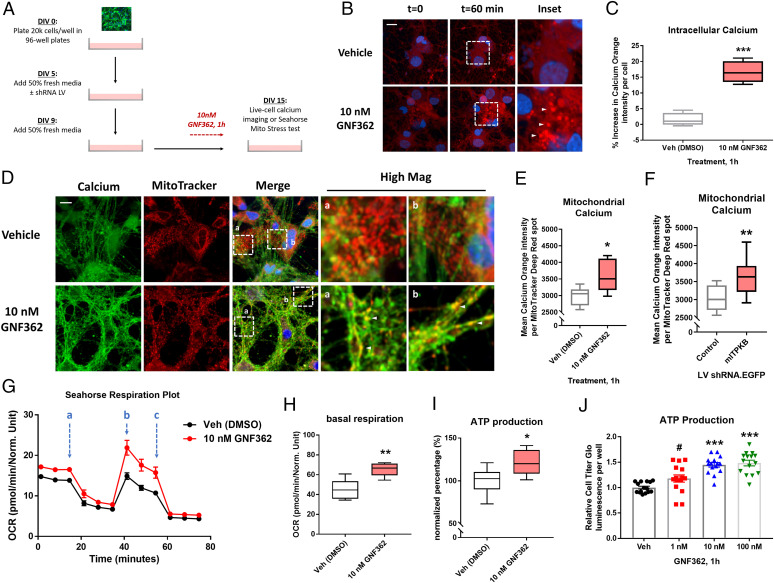
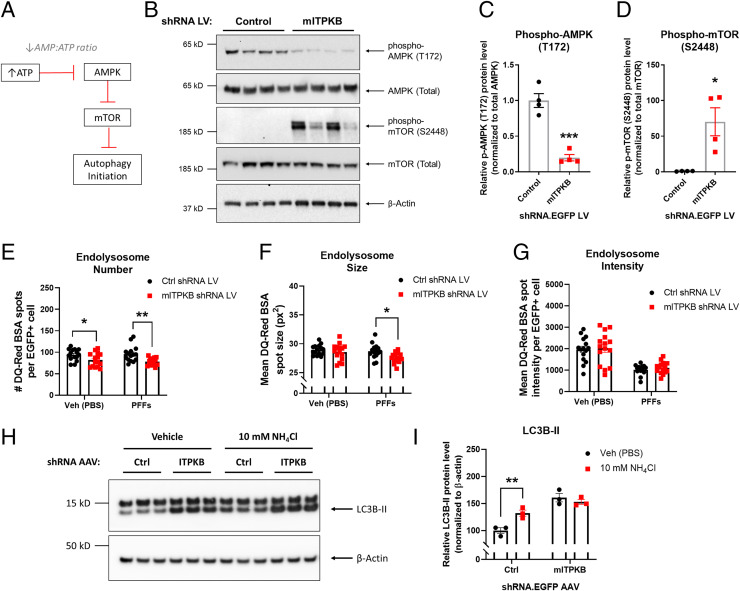
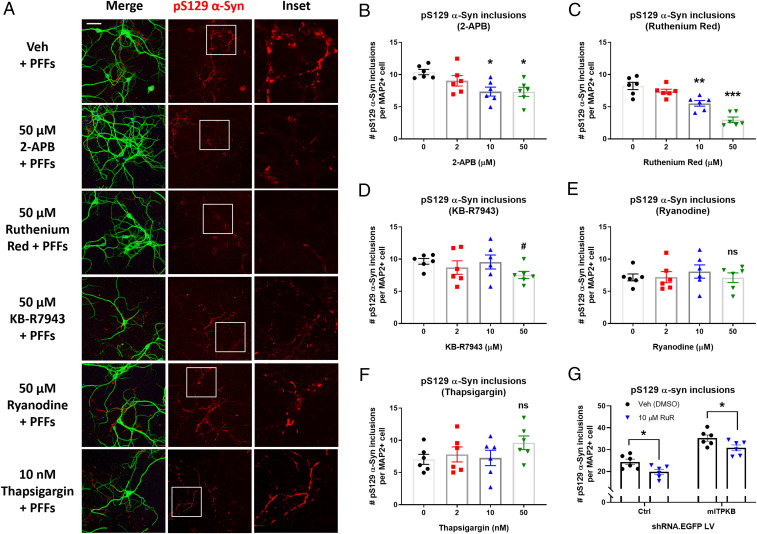
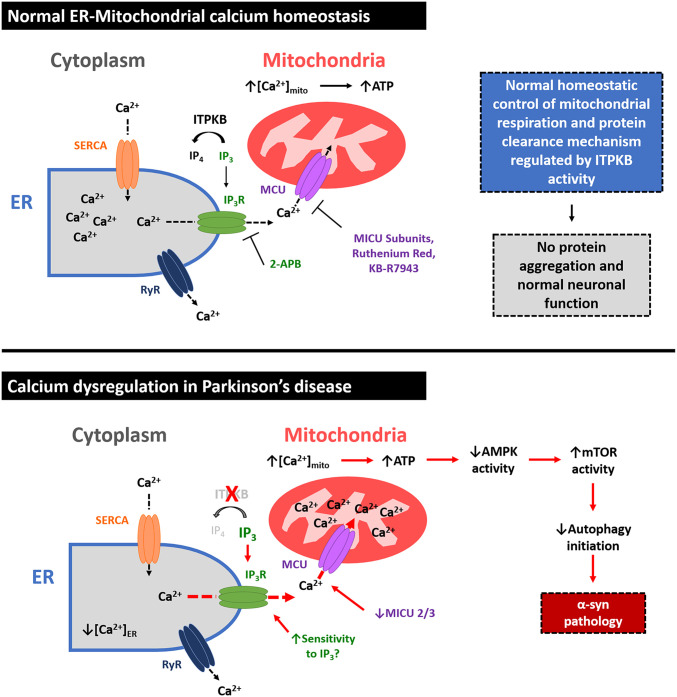
Inositol-1,4,5-triphosphate (IP3) kinase B (ITPKB) is a ubiquitously expressed lipid kinase that inactivates IP3, a secondary messenger that stimulates calcium release from the endoplasmic reticulum (ER). Genome-wide association studies have identified common variants in the ITPKB gene locus associated with reduced risk of sporadic Parkinson's disease (PD). Here, we investigate whether ITPKB activity or expression level impacts PD phenotypes in cellular and animal models. In primary neurons, knockdown or pharmacological inhibition of ITPKB increased levels of phosphorylated, insoluble α-synuclein pathology following treatment with α-synuclein preformed fibrils (PFFs). Conversely, ITPKB overexpression reduced PFF-induced α-synuclein aggregation. We also demonstrate that ITPKB inhibition or knockdown increases intracellular calcium levels in neurons, leading to an accumulation of calcium in mitochondria that increases respiration and inhibits the initiation of autophagy, suggesting that ITPKB regulates α-synuclein pathology by inhibiting ER-to-mitochondria calcium transport. Furthermore, the effects of ITPKB on mitochondrial calcium and respiration were prevented by pretreatment with pharmacological inhibitors of the mitochondrial calcium uniporter complex, which was also sufficient to reduce α-synuclein pathology in PFF-treated neurons. Taken together, these results identify ITPKB as a negative regulator of α-synuclein aggregation and highlight modulation of ER-to-mitochondria calcium flux as a therapeutic strategy for the treatment of sporadic PD.
Keywords: Parkinson’s disease; calcium signaling; genetics; mitochondria; α-synuclein.
Copyright © 2021 the Author(s). Published by PNAS.
Conflict of interest statement
Competing interest statement: E.S., C.L.N., D.T.T., E.G., J.W.N., C.M.B., Y.C., K.E.G., N.A.A., G.C.K., E.A.T., J.G., Y.L., A.W., J.A.H., H.R., and W.D.H. are employees of Biogen. D.J.A., L.T.D., and K.-D.H.N. were employed at Biogen at the time the work was performed but are now employed at AbbVie, Alnylam, and Gemini Therapeutics, respectively.
Figures
References
-
- Goedert M., Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 349, 1255555 (2015). - PubMed
-
- Bonifati V., Genetics of Parkinson’s disease. Minerva Med. 96, 175–186 (2005). - PubMed
-
- Duyckaerts C., et al. , Seeding and propagation of lesions in neurodegenerative diseases: A new paradigm. Bull. Acad Natl. Med. 199, 809–819 (2015). - PubMed
-
- Peelaerts W., Bousset L., Baekelandt V., Melki R., ɑ-synuclein strains and seeding in Parkinson’s disease, incidental Lewy body disease, dementia with Lewy bodies and multiple system atrophy: Similarities and differences. Cell Tissue Res. 373, 195–212 (2018). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
