

A class of viral inducer of degradation of the necroptosis adaptor RIPK3 regulates virus-induced inflammation
- PMID: 33444549
- PMCID: PMC7878414
- DOI: 10.1016/j.immuni.2020.11.020
A class of viral inducer of degradation of the necroptosis adaptor RIPK3 regulates virus-induced inflammation
Abstract
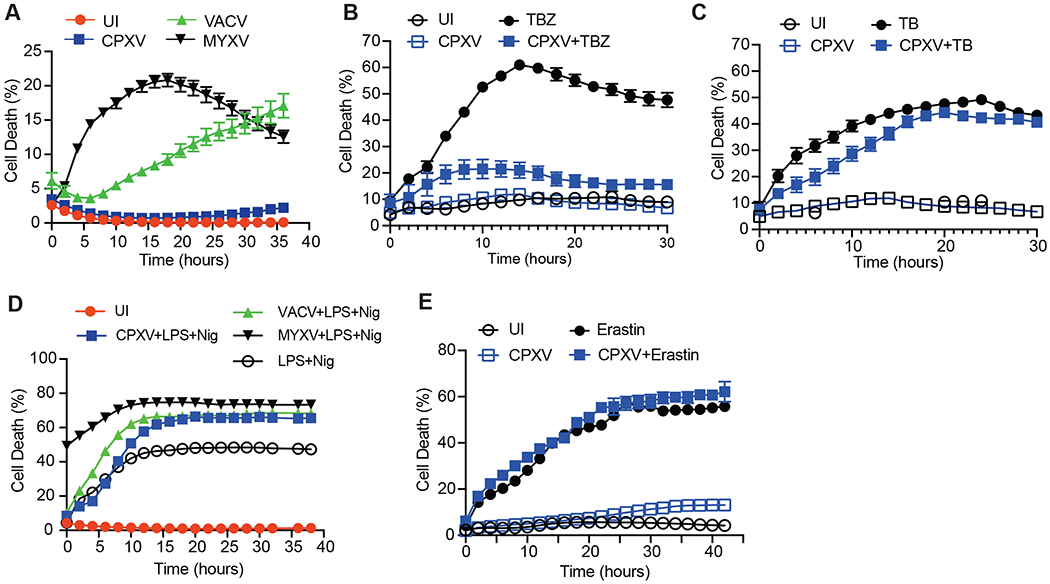
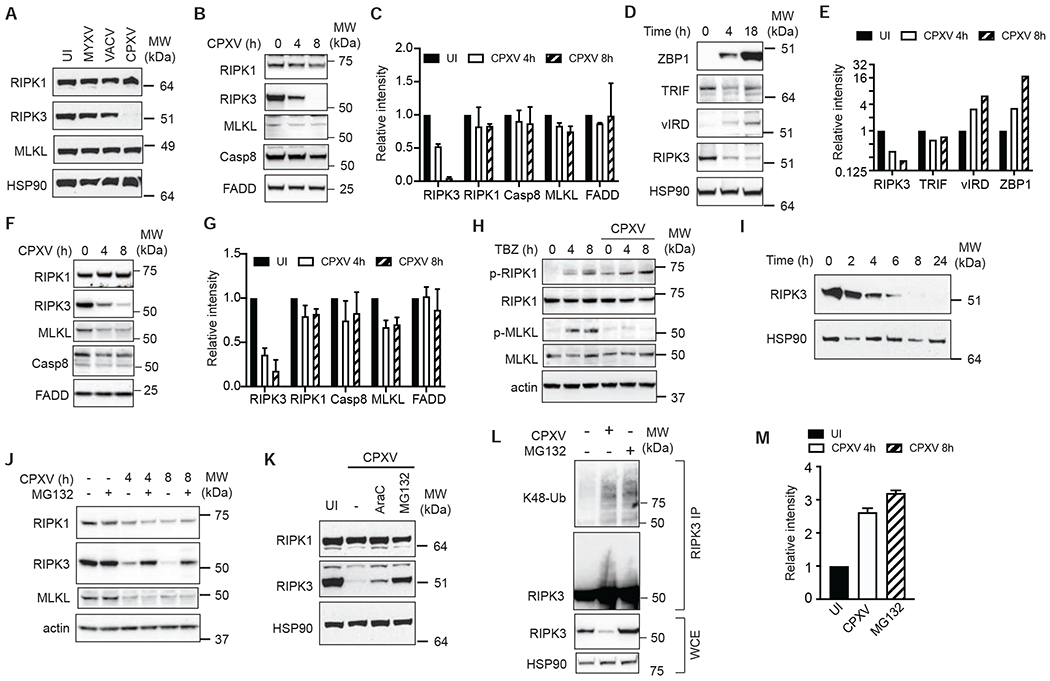
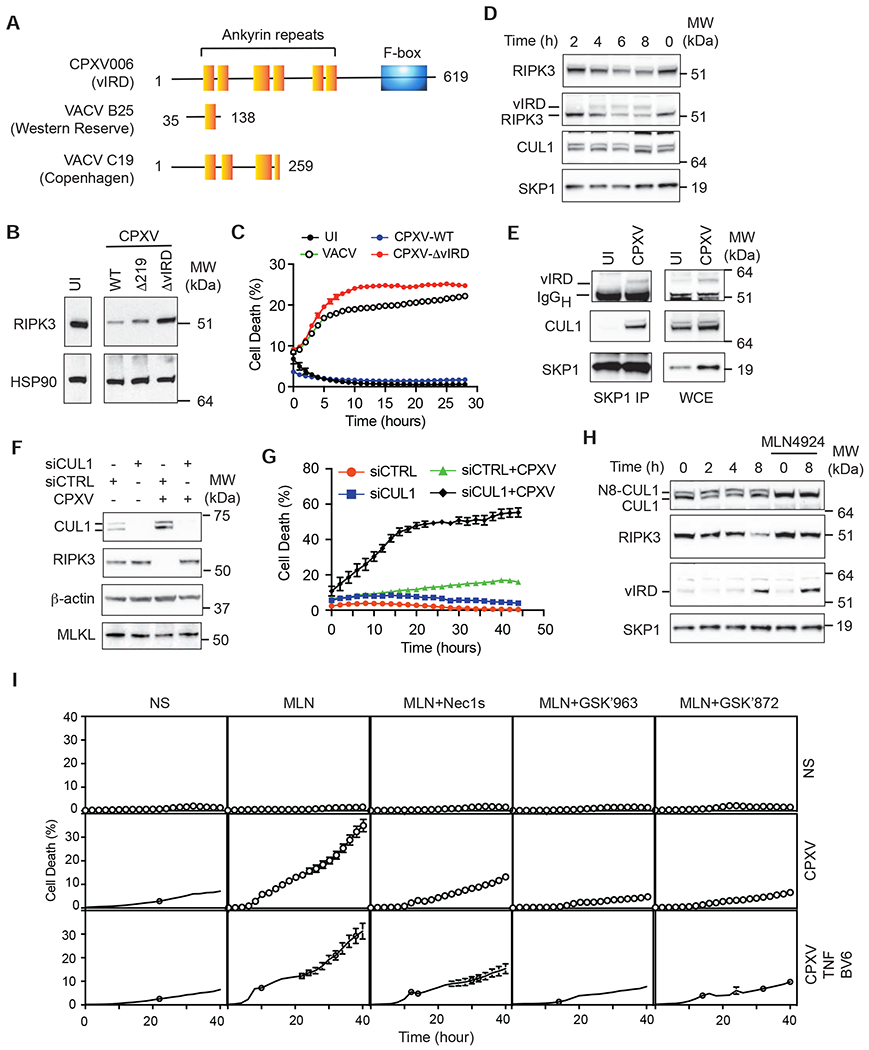
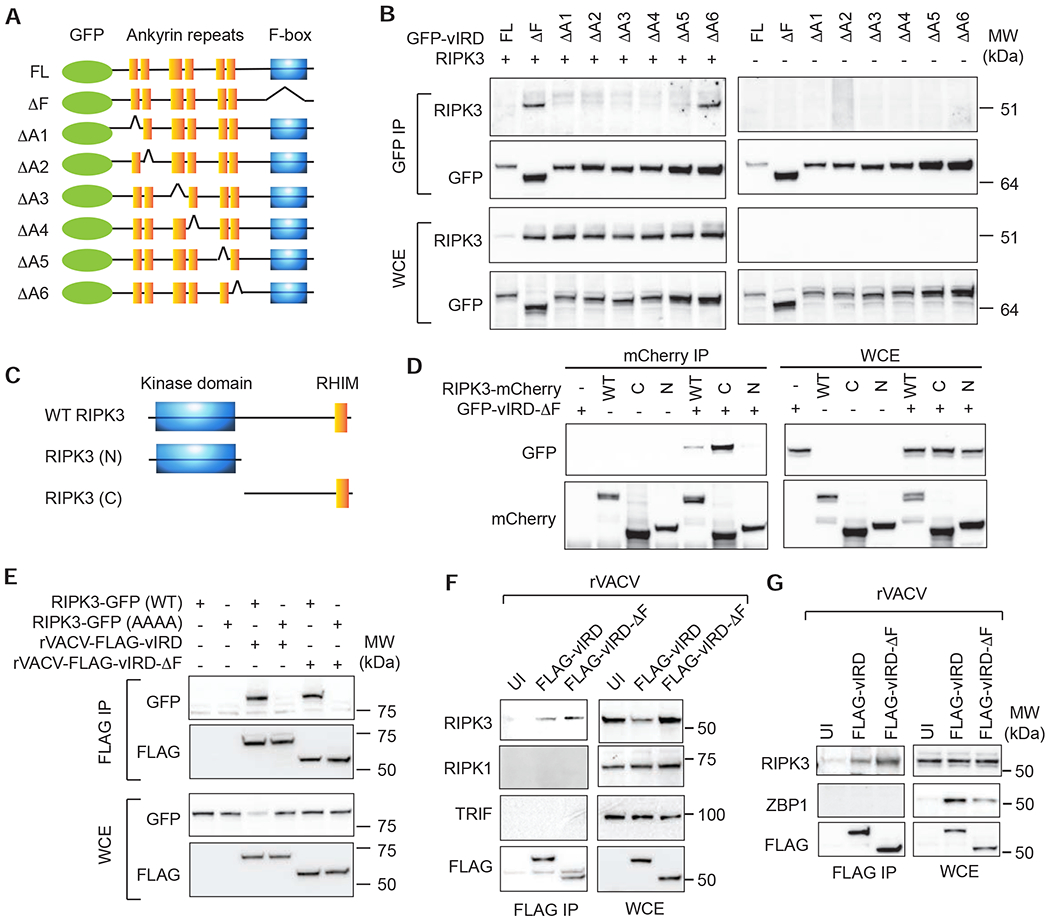
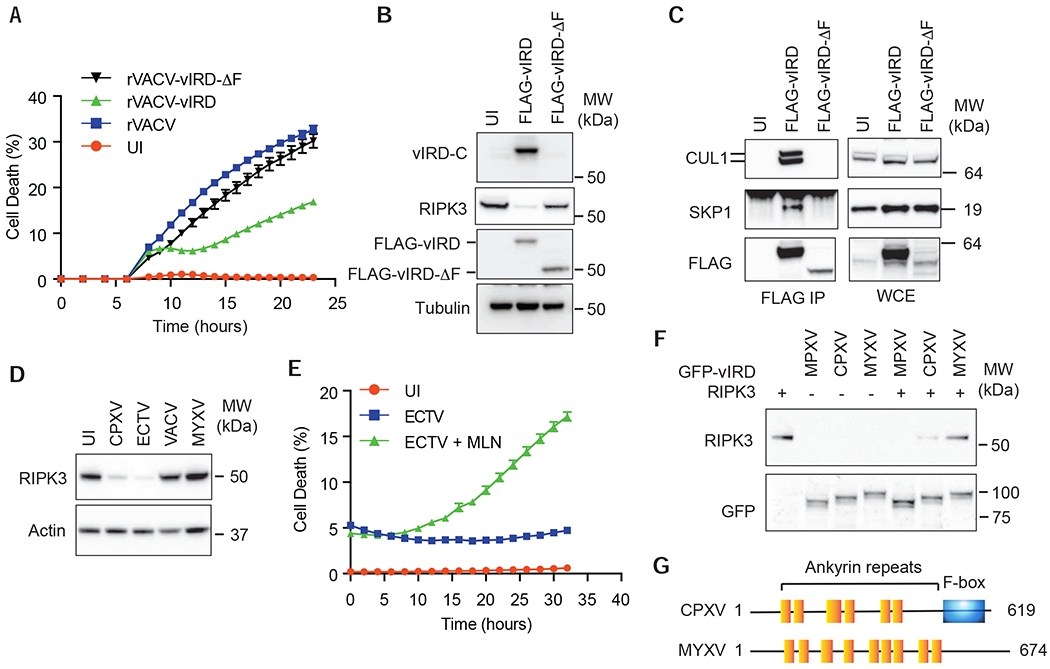
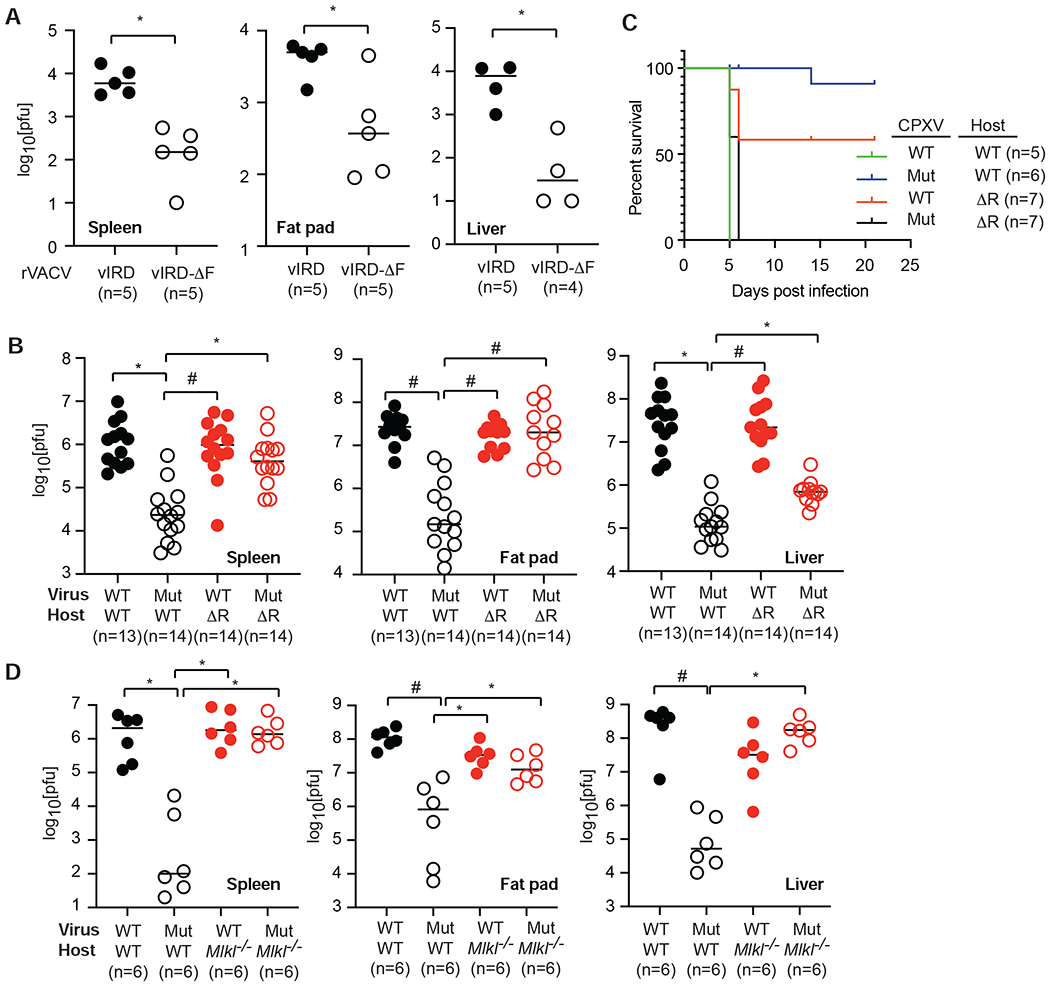
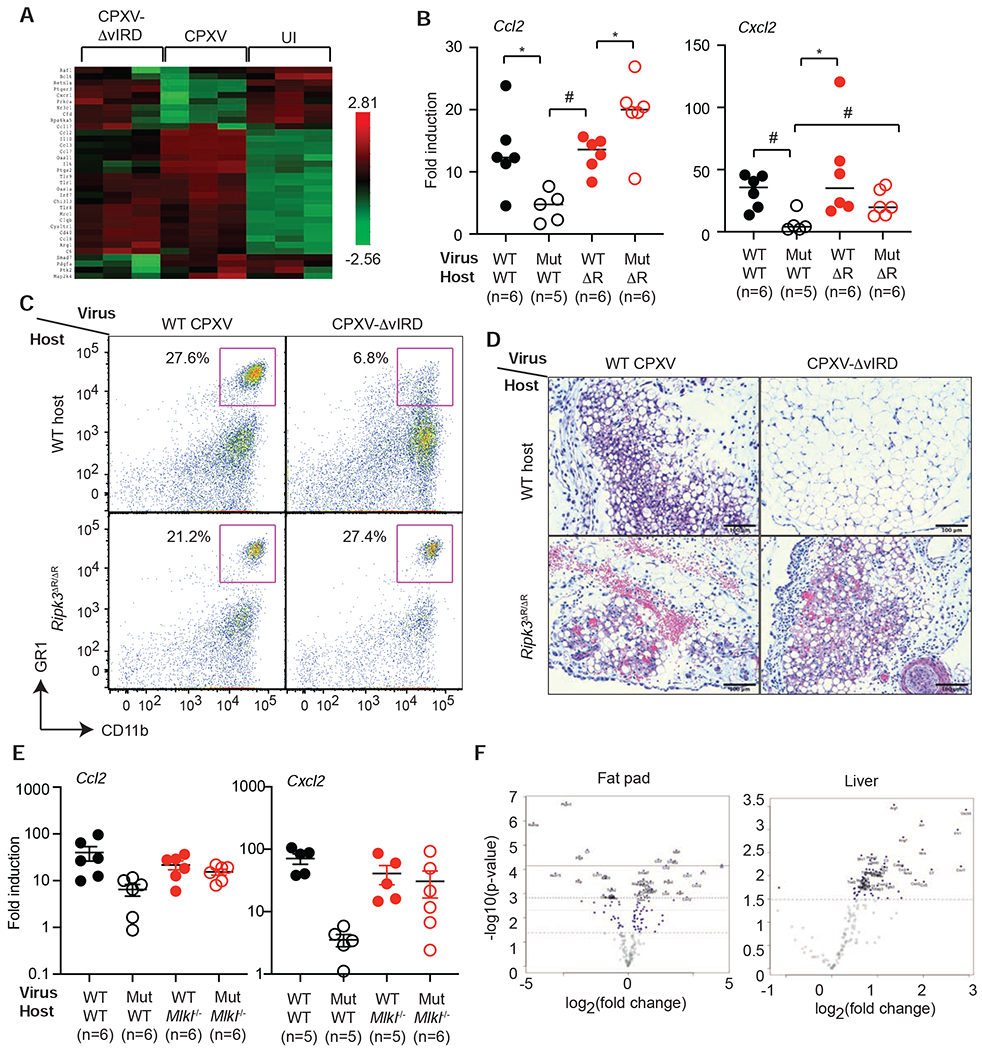
The vaccine strain against smallpox, vaccinia virus (VACV), is highly immunogenic yet causes relatively benign disease. These attributes are believed to be caused by gene loss in VACV. Using a targeted small interfering RNA (siRNA) screen, we identified a viral inhibitor found in cowpox virus (CPXV) and other orthopoxviruses that bound to the host SKP1-Cullin1-F-box (SCF) machinery and the essential necroptosis kinase receptor interacting protein kinase 3 (RIPK3). This "viral inducer of RIPK3 degradation" (vIRD) triggered ubiquitination and proteasome-mediated degradation of RIPK3 and inhibited necroptosis. In contrast to orthopoxviruses, the distantly related leporipoxvirus myxoma virus (MYXV), which infects RIPK3-deficient hosts, lacks a functional vIRD. Introduction of vIRD into VACV, which encodes a truncated and defective vIRD, enhanced viral replication in mice. Deletion of vIRD reduced CPXV-induced inflammation, viral replication, and mortality, which were reversed in RIPK3- and MLKL-deficient mice. Hence, vIRD-RIPK3 drives pathogen-host evolution and regulates virus-induced inflammation and pathogenesis.
Keywords: F-box; RIPK3; TNF; ankyrin repeats; cowpox virus; inflammation; necroptosis; poxvirus; ubiquitination; vaccinia virus.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A U.S. provisional patent application (no. 62/848,255) has been filed for this study.
Figures
References
-
- Blasco R, and Moss B (1995). Selection of recombinant vaccinia viruses on the basis of plaque formation. Gene 158, 157–162. - PubMed
-
- Carpenter EA, Ruby J, and Ramshaw IA (1994). IFN-gamma, TNF, and IL-6 production by vaccinia virus immune spleen cells. An in vitro study. J Immunol 152, 2652–2659. - PubMed
-
- Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, and Lenardo MJ (2003). A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem 278, 51613–51621. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
