

Acute Intermittent Porphyria: An Overview of Therapy Developments and Future Perspectives Focusing on Stabilisation of HMBS and Proteostasis Regulators
- PMID: 33445488
- PMCID: PMC7827610
- DOI: 10.3390/ijms22020675
Acute Intermittent Porphyria: An Overview of Therapy Developments and Future Perspectives Focusing on Stabilisation of HMBS and Proteostasis Regulators
Abstract
Acute intermittent porphyria (AIP) is an autosomal dominant inherited disease with low clinical penetrance, caused by mutations in the hydroxymethylbilane synthase (HMBS) gene, which encodes the third enzyme in the haem biosynthesis pathway. In susceptible HMBS mutation carriers, triggering factors such as hormonal changes and commonly used drugs induce an overproduction and accumulation of toxic haem precursors in the liver. Clinically, this presents as acute attacks characterised by severe abdominal pain and a wide array of neurological and psychiatric symptoms, and, in the long-term setting, the development of primary liver cancer, hypertension and kidney failure. Treatment options are few, and therapies preventing the development of symptomatic disease and long-term complications are non-existent. Here, we provide an overview of the disorder and treatments already in use in clinical practice, in addition to other therapies under development or in the pipeline. We also introduce the pathomechanistic effects of HMBS mutations, and present and discuss emerging therapeutic options based on HMBS stabilisation and the regulation of proteostasis. These are novel mechanistic therapeutic approaches with the potential of prophylactic correction of the disease by totally or partially recovering the enzyme functionality. The present scenario appears promising for upcoming patient-tailored interventions in AIP.
Keywords: acute intermittent porphyria; enzyme intermediates; haem; hydroxymethylbilane synthase; pharmacological chaperones; porphobilinogen deaminase; protein stabilisation; proteostasis regulators; pyrrole chain elongation.
Conflict of interest statement
The authors have filed a patent application for the potential of compound 5- (2-chlorophenyl)methyl]-2-hydroxy-3-nitrobenzaldehyde and 4-chloro-3-nitrophenyl(phenyl)methanone for development of a treatment for acute intermittent porphyria. United Kingdom Patent Application No. 1,916,983.8.
Figures
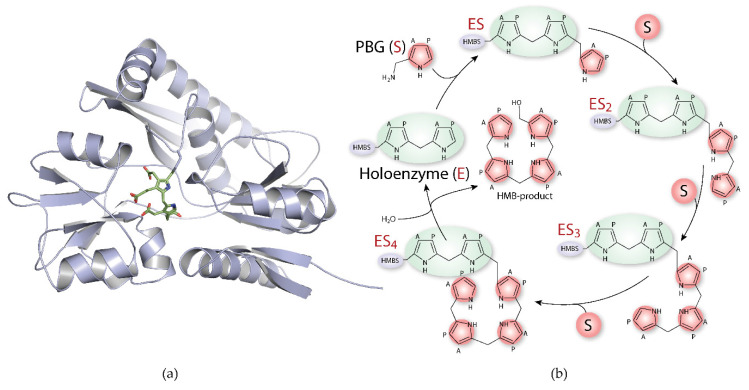
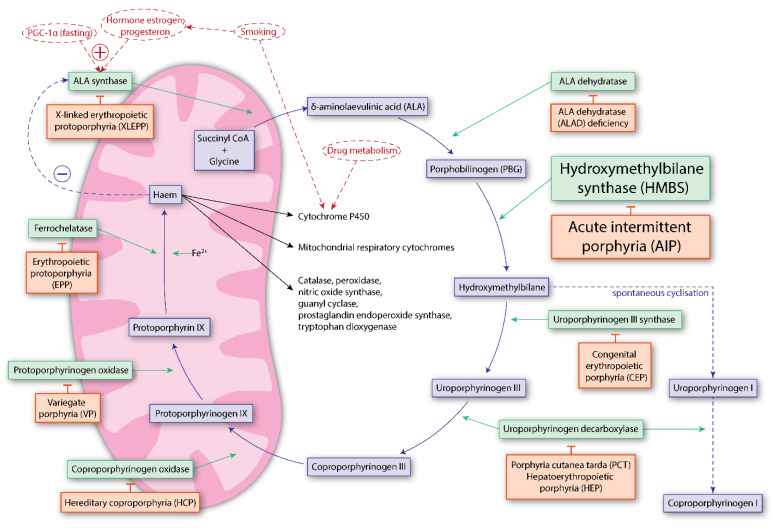
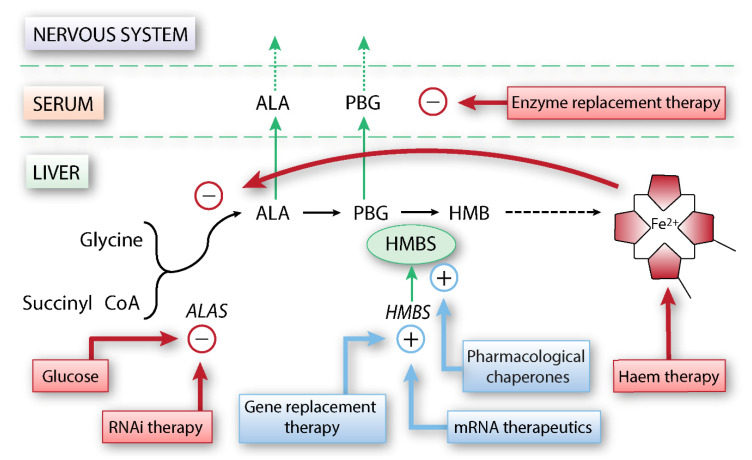
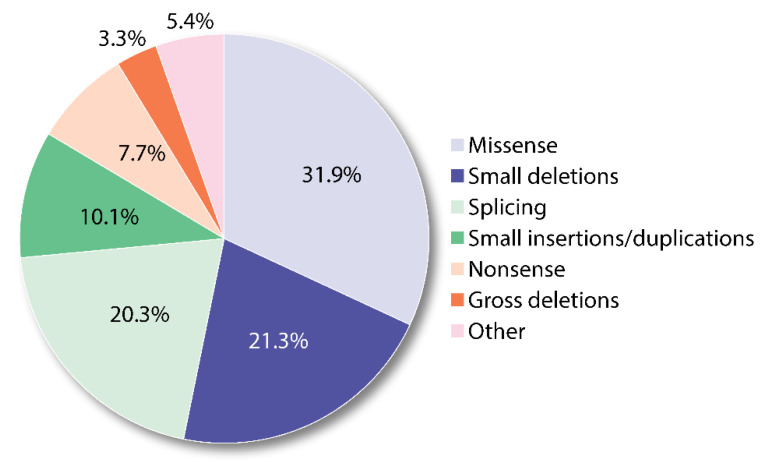
References
-
- Badminton M., Whatley S., Aarsand A.K. Porphyrins and Porphyrias. In: Rifai N., Horvath A.R., Wittwer C., editors. Tietz Textbook of Clinical Chemistry and Molecular Diagnostics. 8th ed. Elsevier; St. Louis, MO, USA: 2018. pp. 776–880.
-
- Pallet N., Mami I., Schmitt C., Karim Z., François A., Rabant M., Nochy D., Gouya L., Deybach J.-C., Xu-Dubois Y., et al. High prevalence of and potential mechanisms for chronic kidney disease in patients with acute intermittent porphyria. Kidney Int. 2015;88:386–395. doi: 10.1038/ki.2015.97. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
