

Bone Marrow Failure Syndromes, Overlapping Diseases with a Common Cytokine Signature
- PMID: 33445786
- PMCID: PMC7828244
- DOI: 10.3390/ijms22020705
Bone Marrow Failure Syndromes, Overlapping Diseases with a Common Cytokine Signature
Abstract
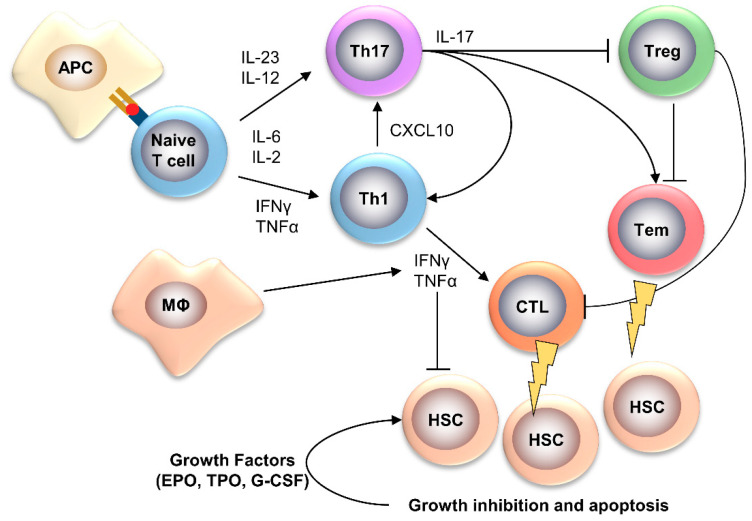
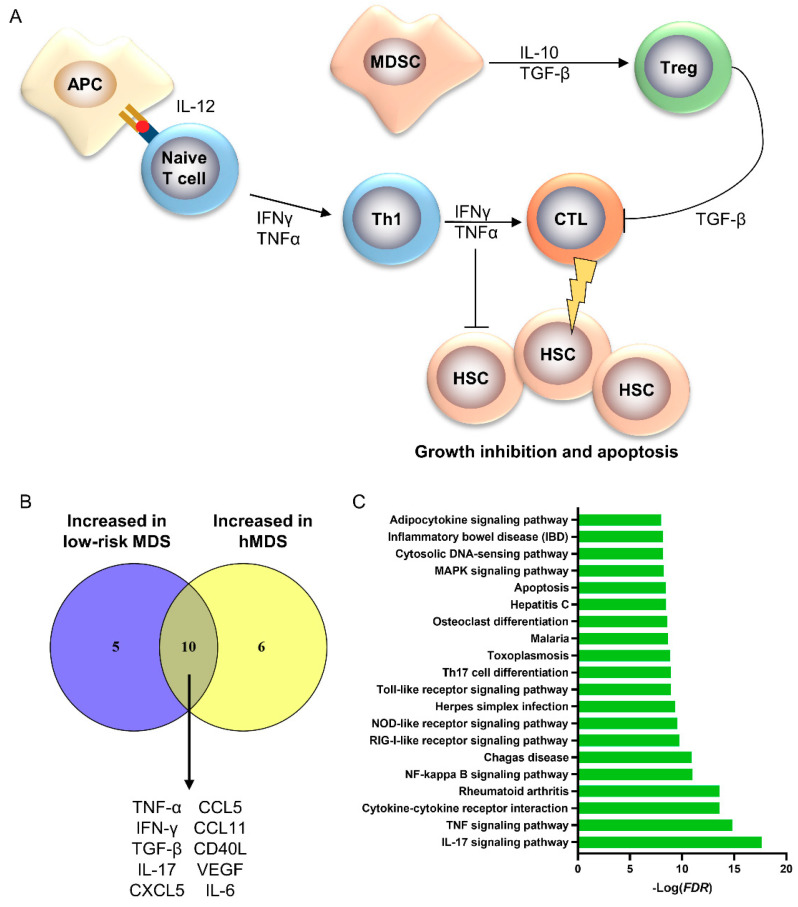
Bone marrow failure (BMF) syndromes are a heterogenous group of non-malignant hematologic diseases characterized by single- or multi-lineage cytopenia(s) with either inherited or acquired pathogenesis. Aberrant T or B cells or innate immune responses are variously involved in the pathophysiology of BMF, and hematological improvement after standard immunosuppressive or anti-complement therapies is the main indirect evidence of the central role of the immune system in BMF development. As part of this immune derangement, pro-inflammatory cytokines play an important role in shaping the immune responses and in sustaining inflammation during marrow failure. In this review, we summarize current knowledge of cytokine signatures in BMF syndromes.
Keywords: aplastic anemia; bone marrow failure syndromes; cytokines; myelodysplastic syndromes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Rodgers G.P., Young N.S. The Bethesda Handbook of Clinical Hematology (English Edition) 4th ed. Lippincott Williams & Wilkins; Philadelphia, PA, USA: 2013.
-
- Park M., Park C.J., Cho Y.W., Jang S., Lee J.H., Lee J.H., Lee K.H., Lee Y.H. Alterations in the bone marrow microenvironment may elicit defective hematopoiesis: A comparison of aplastic anemia, chronic myeloid leukemia, and normal bone marrow. Exp. Hematol. 2017;45:56–63. doi: 10.1016/j.exphem.2016.09.009. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
