

X-linked dominant RPGR gene mutation in a familial Coats angiomatosis
- PMID: 33446141
- PMCID: PMC7807486
- DOI: 10.1186/s12886-020-01791-5
X-linked dominant RPGR gene mutation in a familial Coats angiomatosis
Abstract
Background: Retinitis Pigmentosa (RP) is the most frequent retinal hereditary disease and every kind of transmission pattern has been described. The genetic etiology of RP is extremely heterogeneous and in the last few years the large application of Next Generation Sequencing (NGS) approaches improved the diagnostic yield, elucidating previously unexplained RP causes and new genotype-phenotype correlations. The objective of this study was to reevaluate a previously reported family affected by Coats'-type RP without genetic diagnosis and to describe the new genetic findings.
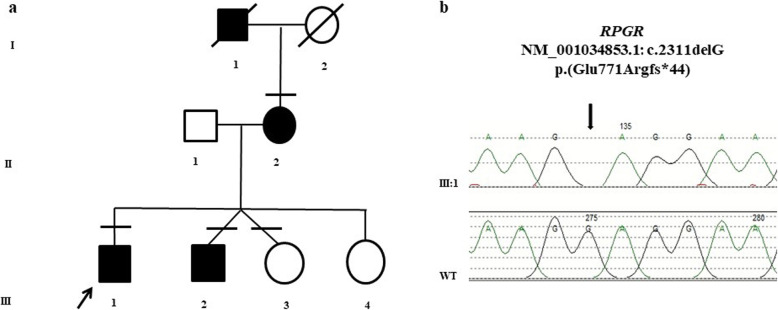
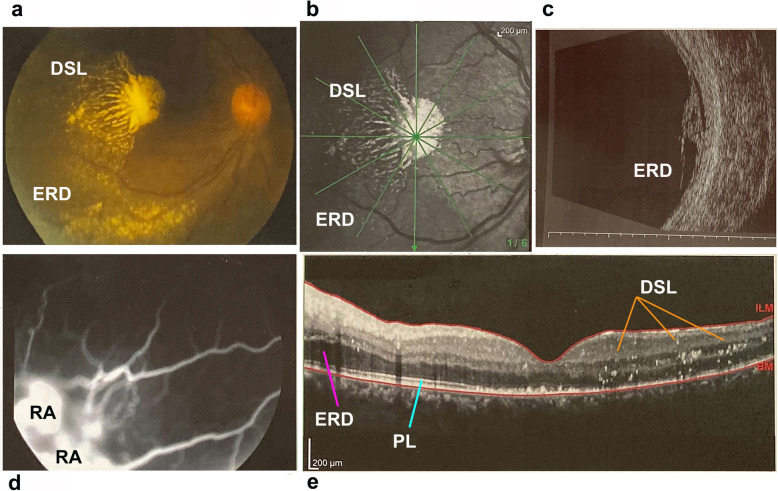
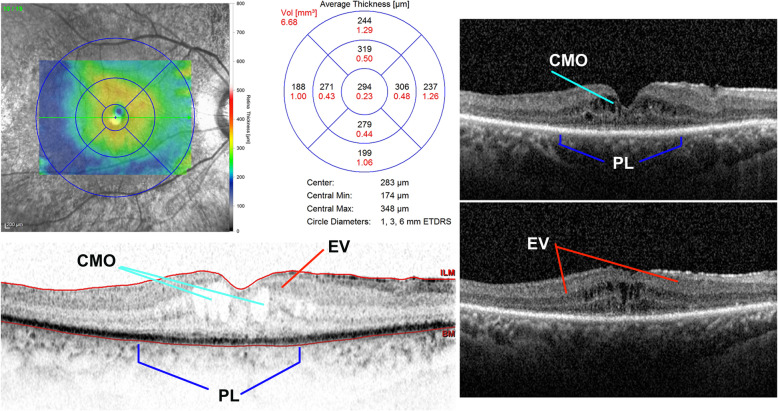
Case presentation: Cohort, prospective, and single-center observational family case. Three individuals of a family, consisting of a mother and four sons, with a Coats phenotype were revaluated after 25 years of clinical follow-up using visual acuity tests, ophthalmoscopy, Goldmann visual field, electroretinography (ERG), and spectral domain-optical coherence tomography (SD-OCT). Specifically, a RP NGS panel was performed on one member of the family and segregation analysis was required for the other affected and unaffected members. NGS analysis disclosed a RPGR (Retinitis Pigmentosa GTPase Regulator) gene truncating variant segregating with the phenotype in all the three affected members. RPGR mutations are reported as causative of an X-linked RP.
Conclusions: This is the first reported family with a Coats'-type RP associated to a RPGR mutation and segregating as a dominant X-linked disease, confirming the hypothesis of the genetic origin of this condition and expanding the phenotypic spectrum of diseases caused by RPGR gene mutations. The Authors suggest RPGR gene screening mutations in patients presenting this phenotype.
Keywords: Case report; Coats vasculopathy; Coats’-type retinitis pigmentosa; Hereditary disease; RPGR- XLRP- Coats’-like retinitis.
Conflict of interest statement
The authors declare that have no competing interests.
Figures
References
-
- Kajiwara Y. Ocular complications of retinitis pigmentosa. Association with Coats’ syndrome. Jpn J Clin Ophthalmol. 1980;34:947–55.
-
- Schmidt D, Faulborn J. Familiares Vorkommen Coats-syndrom kombiniert mit Retinpathia pigmentosa. Klin Monatsbl Augenheilkd. 1972;160(2):158–63. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
