

Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer's disease
- PMID: 33446504
- PMCID: PMC7826333
- DOI: 10.1073/pnas.2017742118
Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer's disease
Abstract
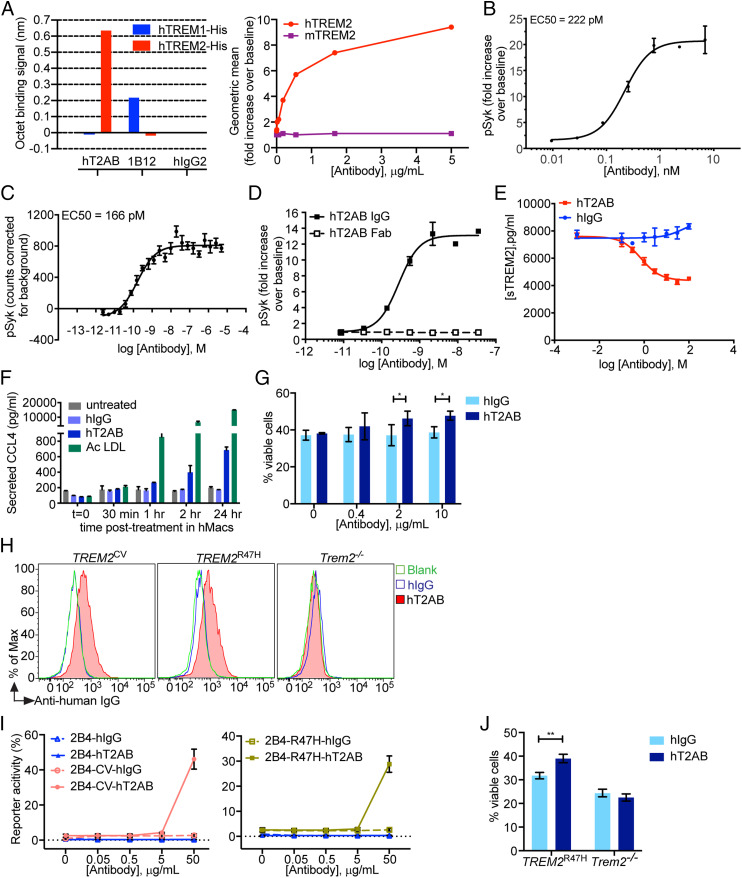
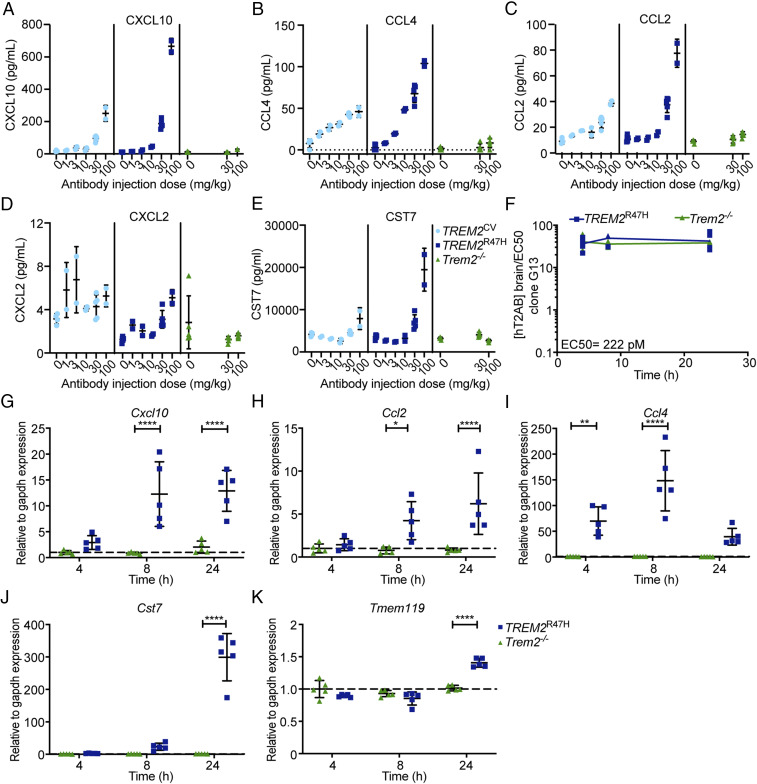
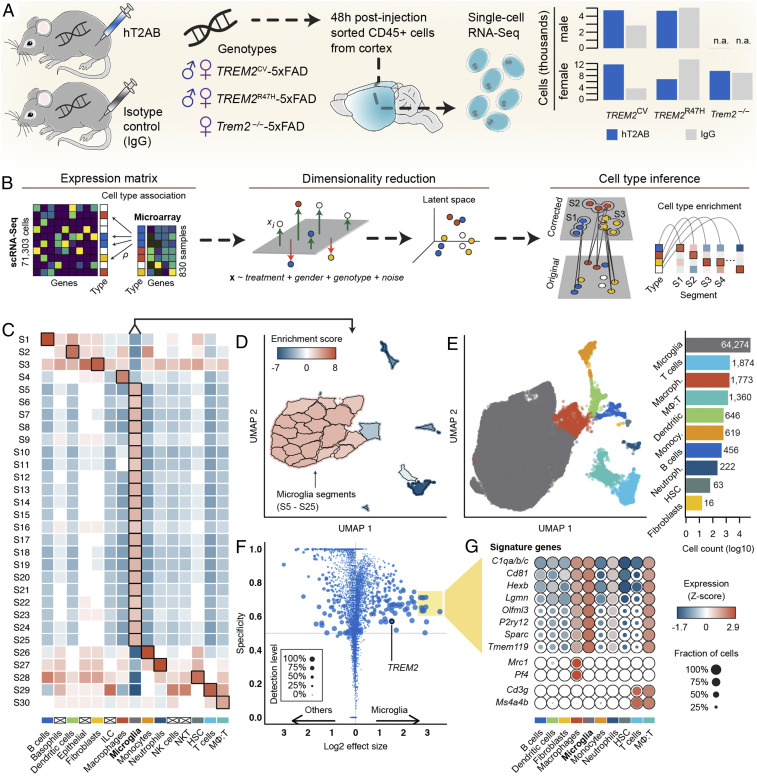
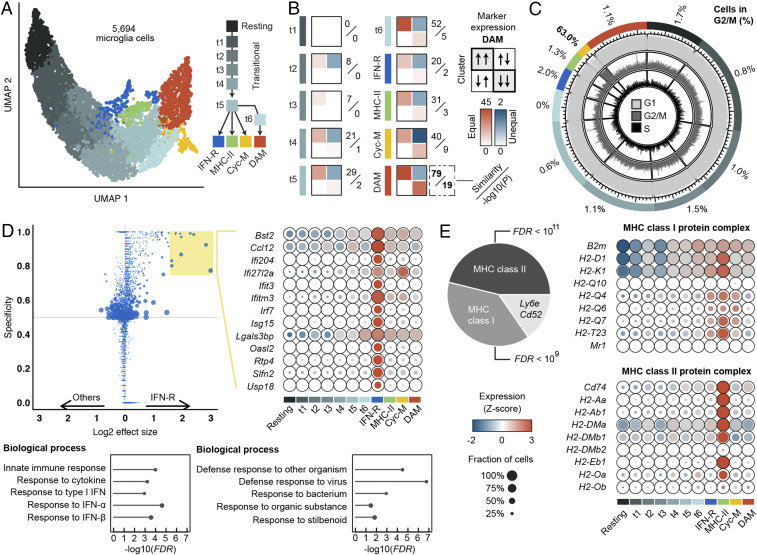
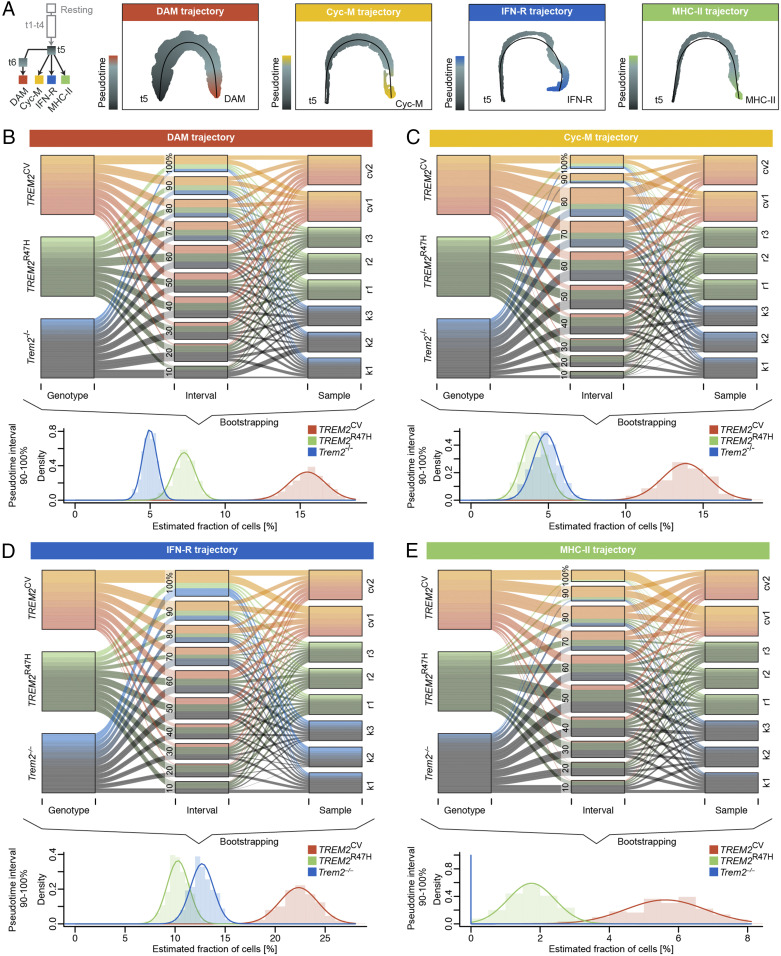
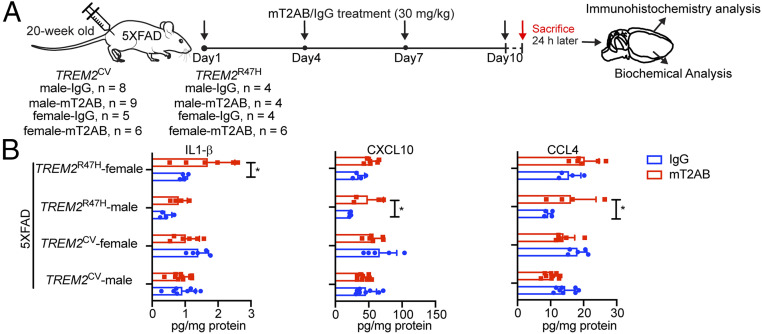
Triggering receptor expressed on myeloid cells 2 (TREM2) sustains microglia response to brain injury stimuli including apoptotic cells, myelin damage, and amyloid β (Aβ). Alzheimer's disease (AD) risk is associated with the TREM2R47H variant, which impairs ligand binding and consequently microglia responses to Aβ pathology. Here, we show that TREM2 engagement by the mAb hT2AB as surrogate ligand activates microglia in 5XFAD transgenic mice that accumulate Aβ and express either the common TREM2 variant (TREM2CV) or TREM2R47H scRNA-seq of microglia from TREM2CV-5XFAD mice treated once with control hIgG1 exposed four distinct trajectories of microglia activation leading to disease-associated (DAM), interferon-responsive (IFN-R), cycling (Cyc-M), and MHC-II expressing (MHC-II) microglia types. All of these were underrepresented in TREM2R47H-5XFAD mice, suggesting that TREM2 ligand engagement is required for microglia activation trajectories. Moreover, Cyc-M and IFN-R microglia were more abundant in female than male TREM2CV-5XFAD mice, likely due to greater Aβ load in female 5XFAD mice. A single systemic injection of hT2AB replenished Cyc-M, IFN-R, and MHC-II pools in TREM2R47H-5XFAD mice. In TREM2CV-5XFAD mice, however, hT2AB brought the representation of male Cyc-M and IFN-R microglia closer to that of females, in which these trajectories had already reached maximum capacity. Moreover, hT2AB induced shifts in gene expression patterns in all microglial pools without affecting representation. Repeated treatment with a murinized hT2AB version over 10 d increased chemokines brain content in TREM2R47H-5XFAD mice, consistent with microglia expansion. Thus, the impact of hT2AB on microglia is shaped by the extent of TREM2 endogenous ligand engagement and basal microglia activation.
Keywords: Alzheimer’s disease; TREM2; amyloid beta; microglia; monoclonal antibody.
Conflict of interest statement
Competing interest statement: D.C.E., Z.S., L.G., R.C., D.Y., D.W., P.R., J.B., S.R., D.C., P.L., S.S., S.A.H., I.N.F., and M.v.L.C. are current employees or were past Amgen employees at the time when the experiments were performed. S.S. is Vice President of Nura Bio Inc. M.C. received research support from Amgen and serves on the Scientific Advisory Board of Vigil Neuroscience Inc.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
