

Differentially expressed lncRNAs in liver tissues of TX mice with hepatolenticular degeneration
- PMID: 33446761
- PMCID: PMC7809420
- DOI: 10.1038/s41598-020-80635-0
Differentially expressed lncRNAs in liver tissues of TX mice with hepatolenticular degeneration
Abstract
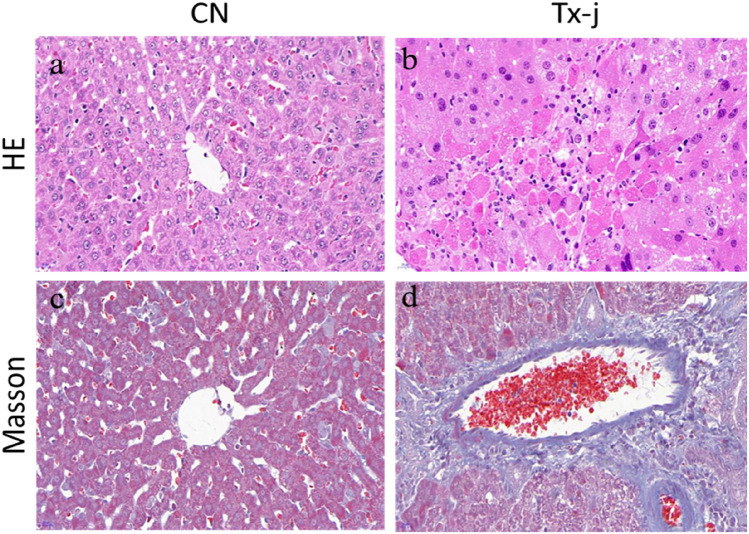
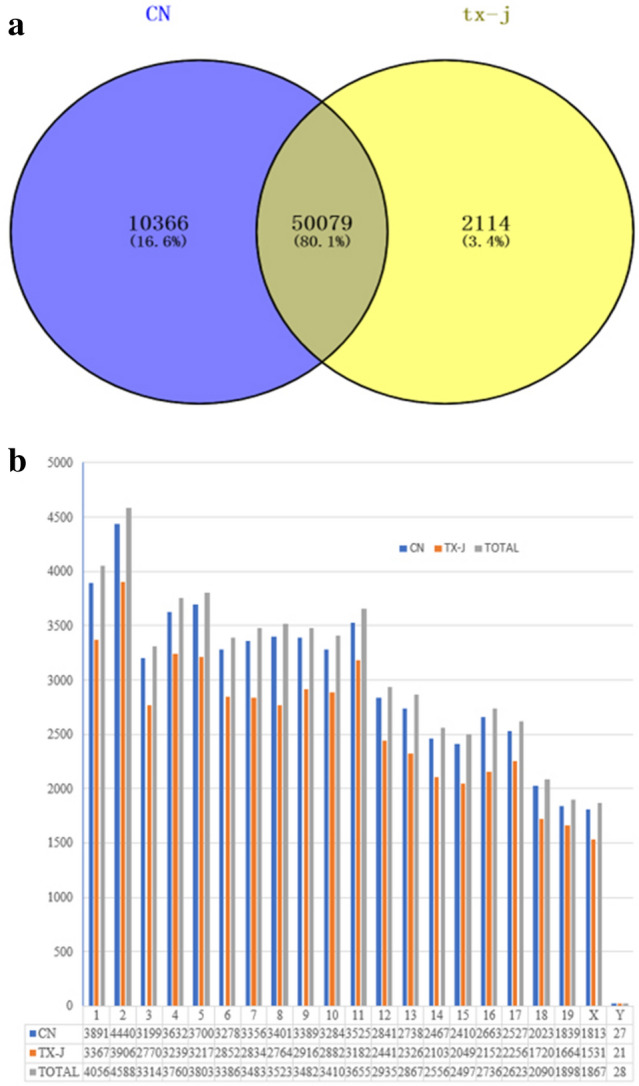
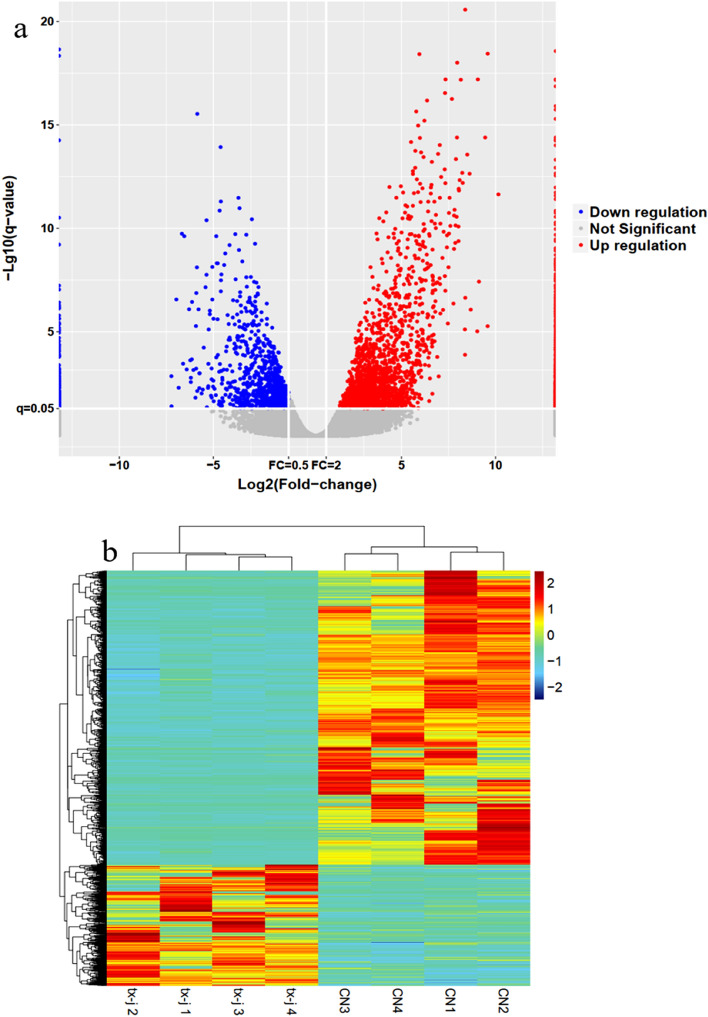
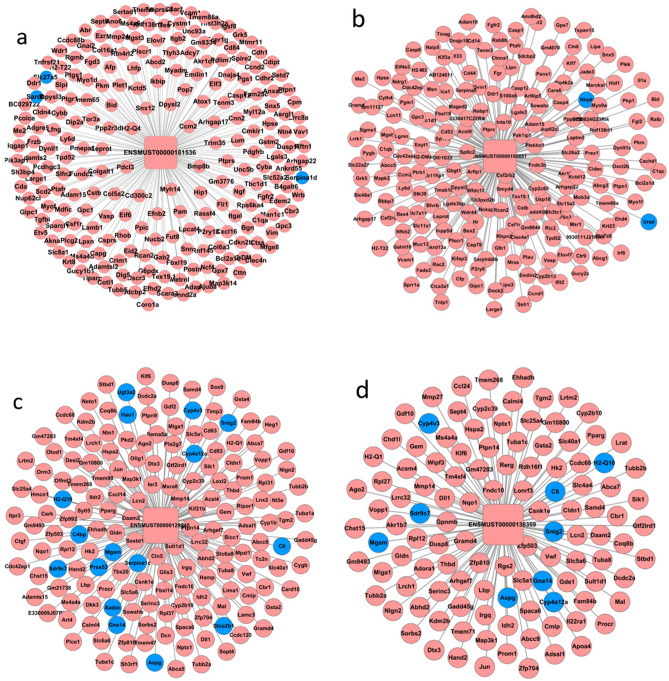
Wilson's Disease (WD), an ATP7B-mutated inherited disease that affects copper transport, is characterised by liver and nervous system manifestations. Long non-coding (ln-c) RNAs are widely involved in almost all physiological and pathological processes in the body, and are associated with numerous diseases. The present study aimed to elucidate the lncRNA-mRNA regulation network in a TX WD mouse model using RNA sequencing (RNA-seq). lncRNA expression profiles were screened using RNA-seq and real-time polymerase chain reaction, and differentially expressed lncRNAs and mRNAs were identified. To analyse the biological functions and pathways for the differentially expressed mRNAs, gene ontology and pathway enrichment analyses were performed. A significantly correlated lncRNA-mRNA relationship pair was calculated by CNC analysis to construct differential lncRNA and mRNA co-expression networks. A total of 2564 significantly up-regulated and 1052 down-regulated lncRNAs, and 1576 up-regulated and 297 down-regulated mRNAs, were identified. These genes were found to be associated with key processes such as apoptosis, and KEGG analysis revealed enrichment in the drug metabolism-cytochrome P450 pathway, PPAR signalling pathway, Notch signalling pathway, and MAPK signalling pathway. The identified differential lncRNAs may be involved in the pathogenesis and development of WD liver injury.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
