

Potential repurposing of four FDA approved compounds with antiplasmodial activity identified through proteome scale computational drug discovery and in vitro assay
- PMID: 33446838
- PMCID: PMC7809352
- DOI: 10.1038/s41598-020-80722-2
Potential repurposing of four FDA approved compounds with antiplasmodial activity identified through proteome scale computational drug discovery and in vitro assay
Abstract
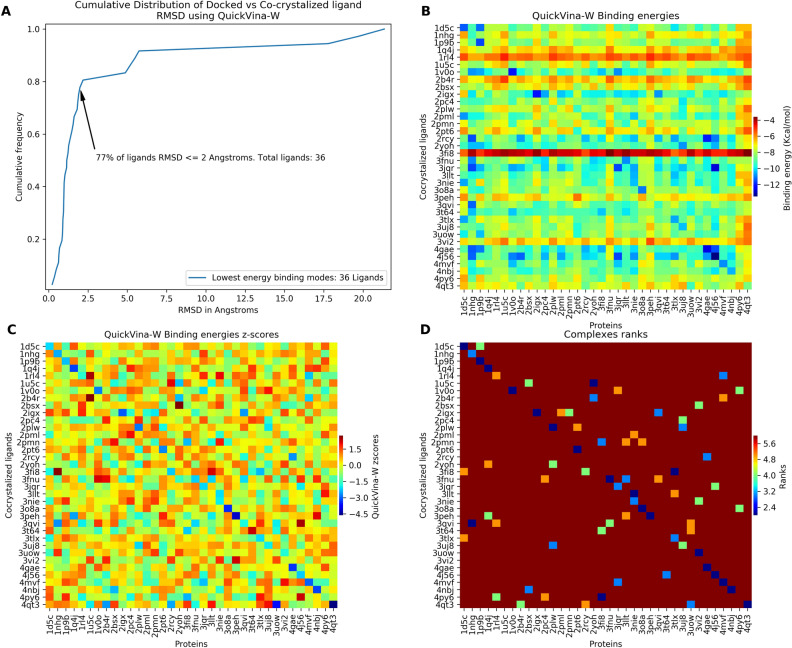
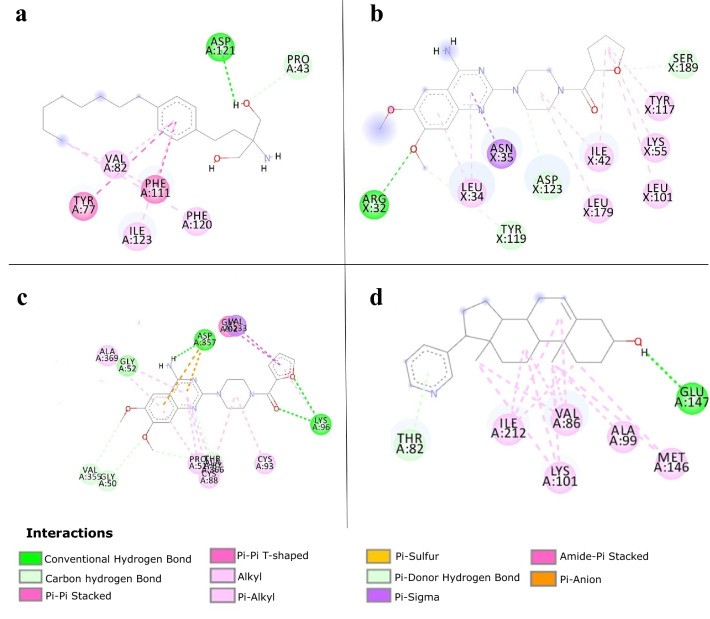
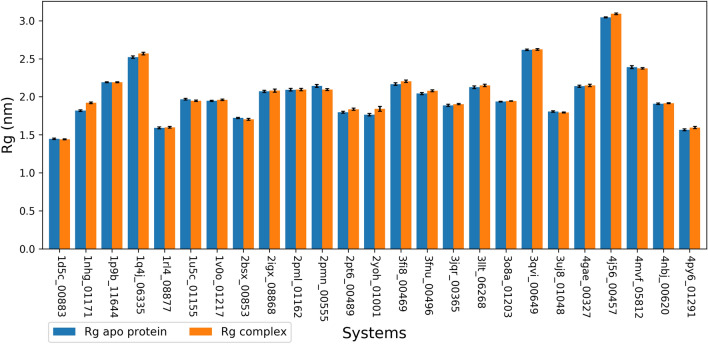
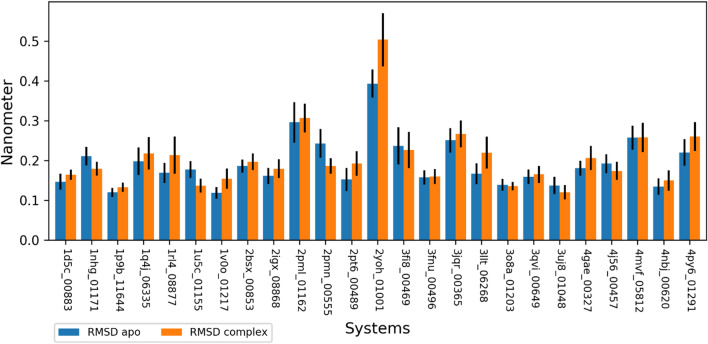
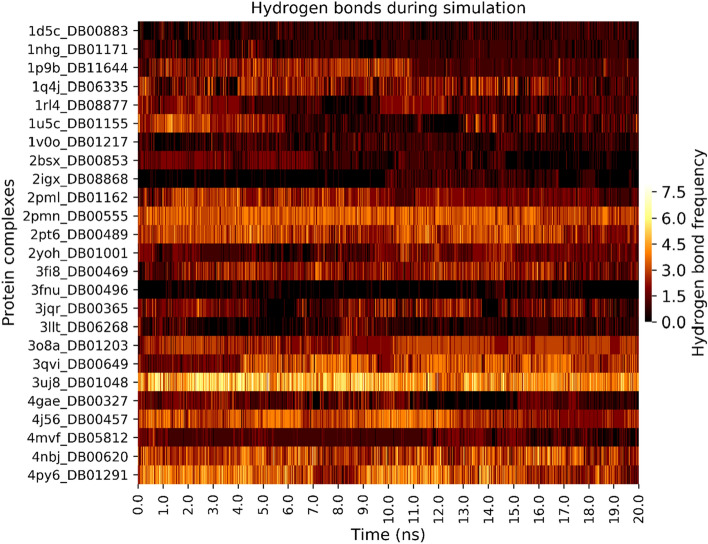
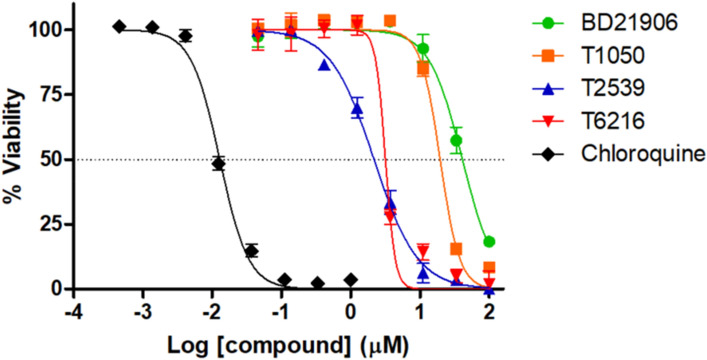
Malaria elimination can benefit from time and cost-efficient approaches for antimalarials such as drug repurposing. In this work, 796 DrugBank compounds were screened against 36 Plasmodium falciparum targets using QuickVina-W. Hits were selected after rescoring using GRaph Interaction Matching (GRIM) and ligand efficiency metrics: surface efficiency index (SEI), binding efficiency index (BEI) and lipophilic efficiency (LipE). They were further evaluated in Molecular dynamics (MD). Twenty-five protein-ligand complexes were finally retained from the 28,656 (36 × 796) dockings. Hit GRIM scores (0.58 to 0.78) showed their molecular interaction similarity to co-crystallized ligands. Minimum LipE (3), SEI (23) and BEI (7) were in at least acceptable thresholds for hits. Binding energies ranged from -6 to -11 kcal/mol. Ligands showed stability in MD simulation with good hydrogen bonding and favorable protein-ligand interactions energy (the poorest being -140.12 kcal/mol). In vitro testing showed 4 active compounds with two having IC50 values in the single-digit μM range.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- World Health Organization. WHO Malaria report 2019. Malaria report 2019https://www.who.int/publications-detail/world-malaria-report-2019 (2019).
-
- Lunev, S., Batista, F. A., Bosch, S. S., Wrenger, C. & Groves, M. R. Identification and Validation of Novel Drug Targets for the Treatment of Plasmodium falciparum Malaria: New Insights. Current Topics in Malaria (InTech, 2016). 10.5772/65659.
-
- Vora P, Somani R, Jain M. Drug repositioning: an approach for drug discovery. Mini. Rev. Org. Chem. 2016;13:363–376. doi: 10.2174/1570193X13666160728121823. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
