

Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer
- PMID: 33450205
- PMCID: PMC8635244
- DOI: 10.1016/j.cell.2020.12.031
Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer
Abstract
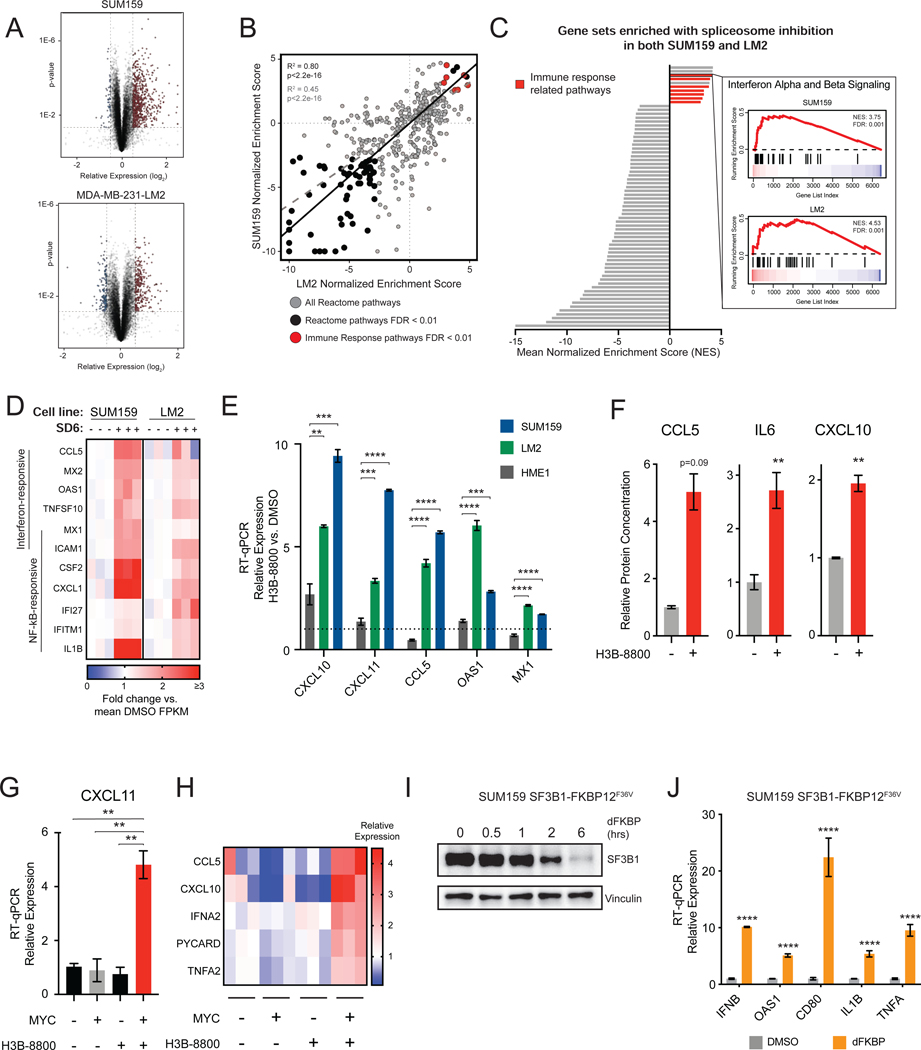
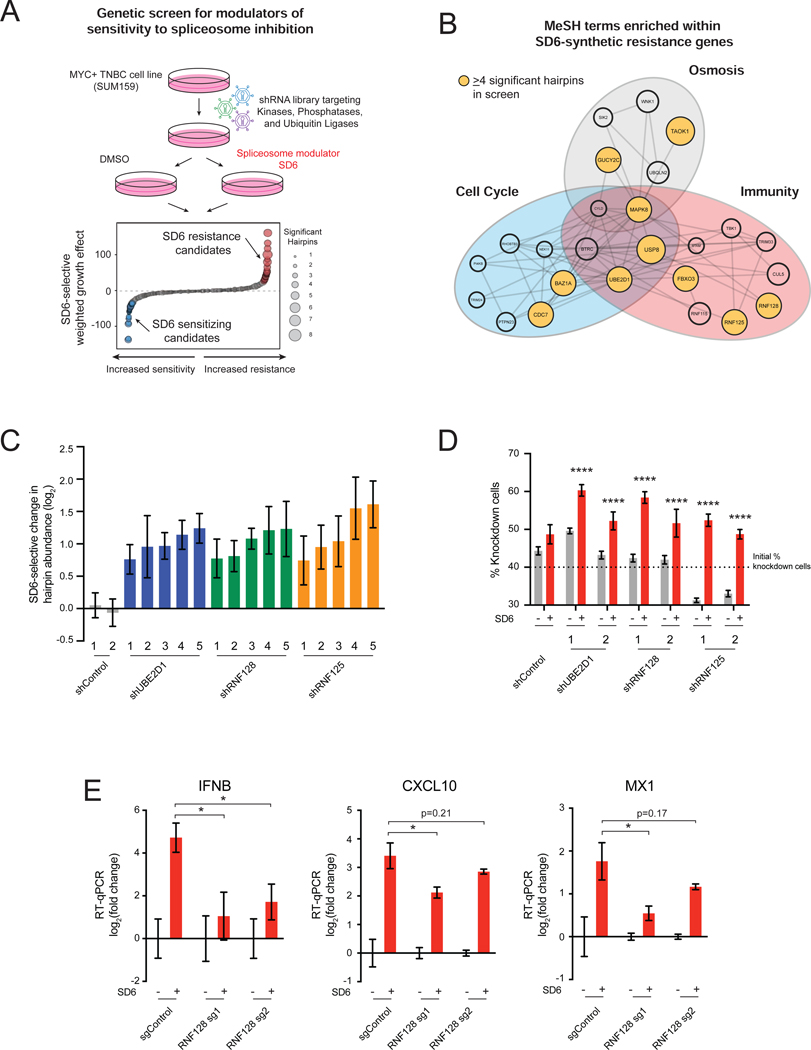
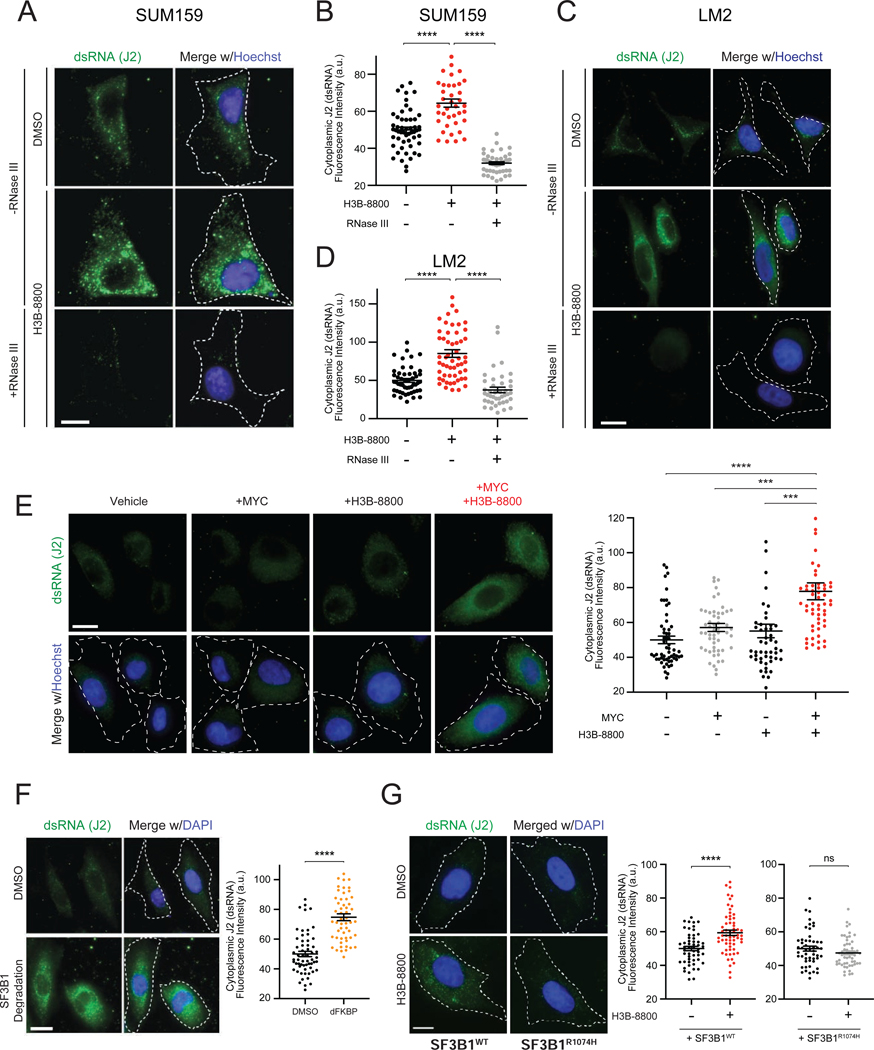
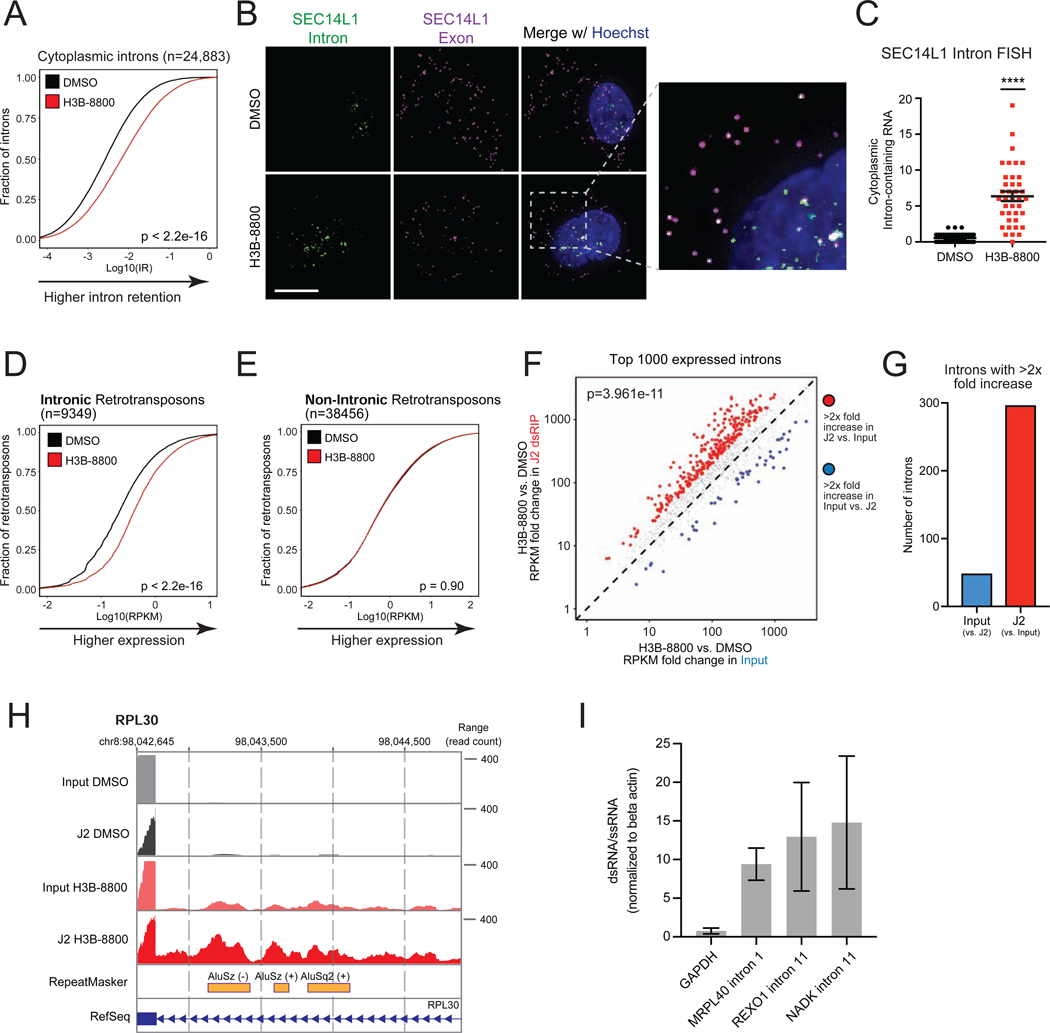
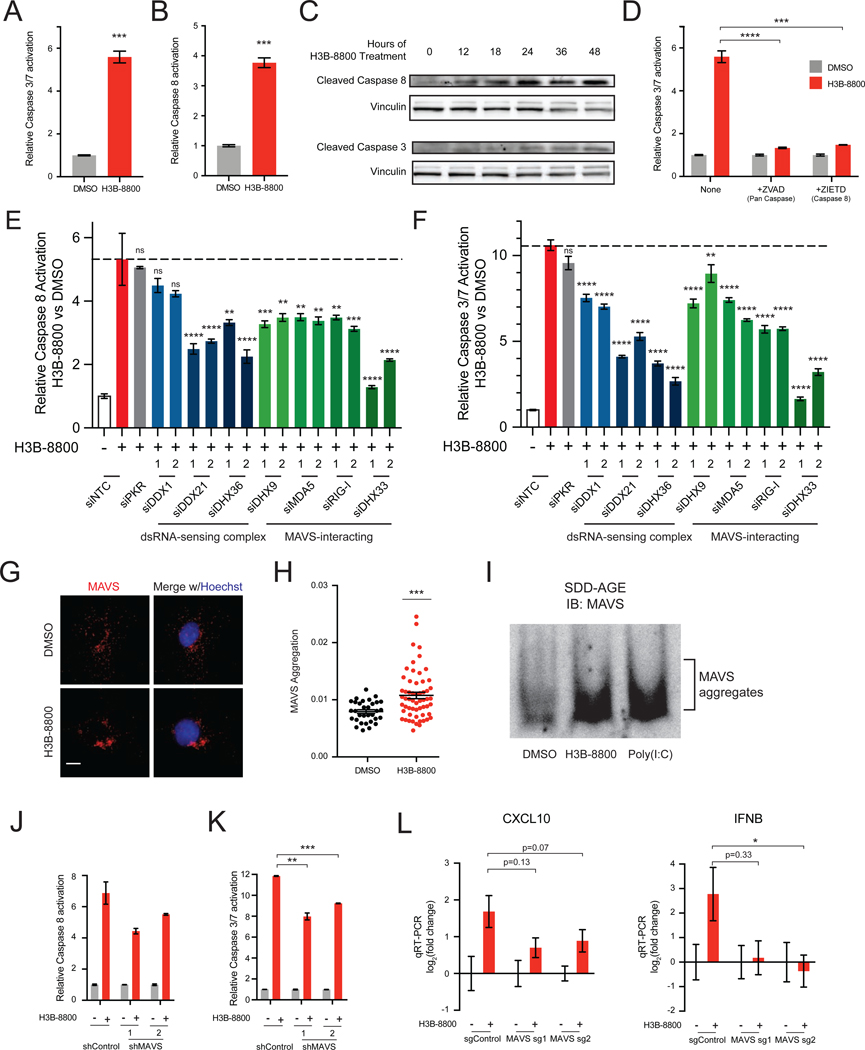
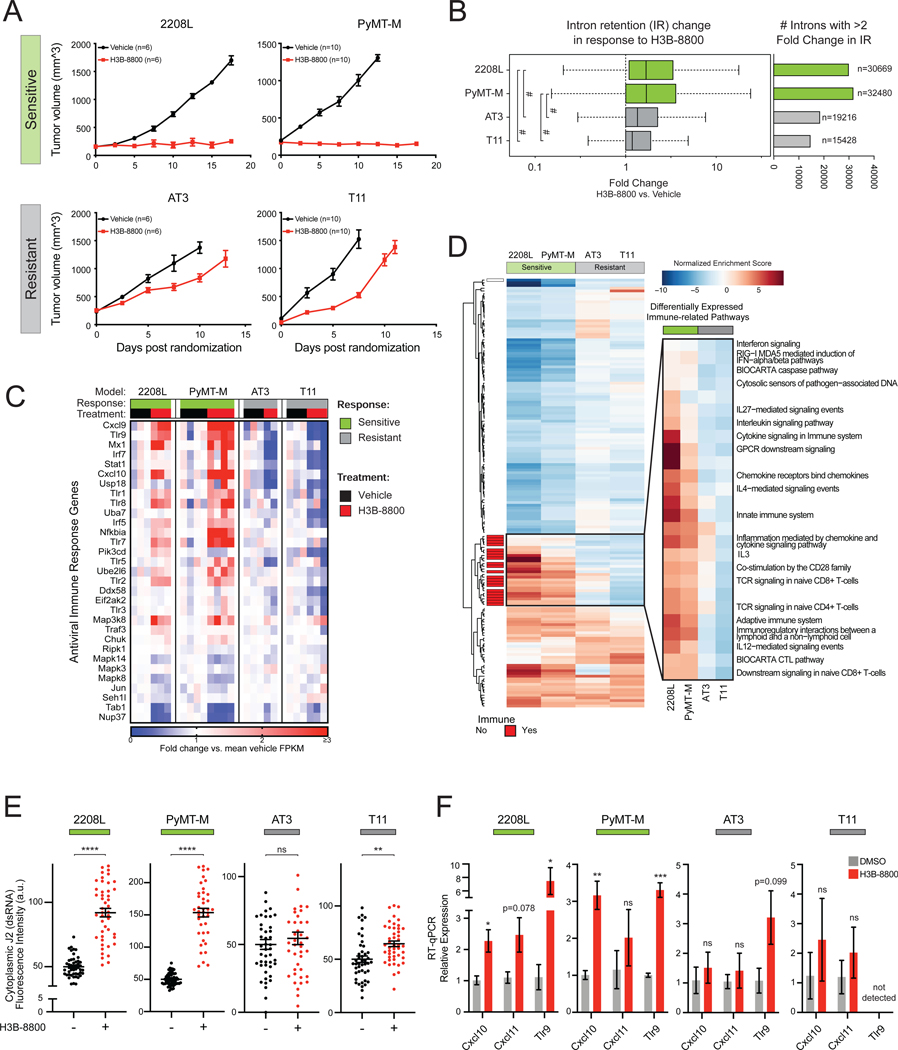
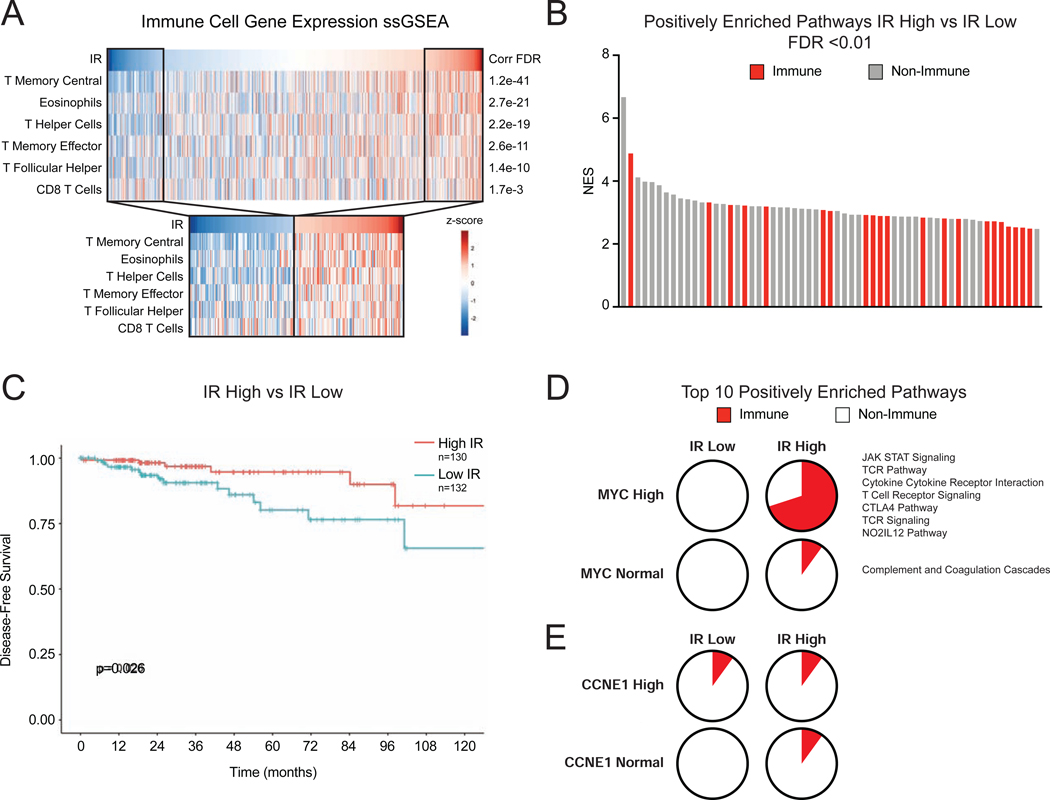
Many oncogenic insults deregulate RNA splicing, often leading to hypersensitivity of tumors to spliceosome-targeted therapies (STTs). However, the mechanisms by which STTs selectively kill cancers remain largely unknown. Herein, we discover that mis-spliced RNA itself is a molecular trigger for tumor killing through viral mimicry. In MYC-driven triple-negative breast cancer, STTs cause widespread cytoplasmic accumulation of mis-spliced mRNAs, many of which form double-stranded structures. Double-stranded RNA (dsRNA)-binding proteins recognize these endogenous dsRNAs, triggering antiviral signaling and extrinsic apoptosis. In immune-competent models of breast cancer, STTs cause tumor cell-intrinsic antiviral signaling, downstream adaptive immune signaling, and tumor cell death. Furthermore, RNA mis-splicing in human breast cancers correlates with innate and adaptive immune signatures, especially in MYC-amplified tumors that are typically immune cold. These findings indicate that dsRNA-sensing pathways respond to global aberrations of RNA splicing in cancer and provoke the hypothesis that STTs may provide unexplored strategies to activate anti-tumor immune pathways.
Keywords: MYC; RNA splicing in cancer; anti-cancer immunity; antiviral immunity; double-stranded RNA; oncogenic stress; spliceosome-targeted therapies; triple-negative breast cancer; viral mimicry.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Comment in
-
Spliceosome-Targeted Therapies Induce dsRNA Responses.Immunity. 2021 Jan 12;54(1):11-13. doi: 10.1016/j.immuni.2020.12.012. Immunity. 2021. PMID: 33440135
-
RNA splicing meets anti-tumor immunity.Nat Cancer. 2021 Dec;2(12):1287. doi: 10.1038/s43018-021-00309-2. Nat Cancer. 2021. PMID: 35121911 No abstract available.
References
-
- Al-Kuraya K, Schraml P, Torhorst J, Tapia C, Zaharieva B, Novotny H, Spichtin H, Maurer R, Mirlacher M, Köchli O, et al., 2004. Prognostic relevance of gene amplifications and coamplifications in breast cancer. Cancer Res. 64, 8534–8540. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
