

A CRISPR/Cas9-Engineered ARID1A-Deficient Human Gastric Cancer Organoid Model Reveals Essential and Nonessential Modes of Oncogenic Transformation
- PMID: 33451982
- PMCID: PMC8346515
- DOI: 10.1158/2159-8290.CD-20-1109
A CRISPR/Cas9-Engineered ARID1A-Deficient Human Gastric Cancer Organoid Model Reveals Essential and Nonessential Modes of Oncogenic Transformation
Abstract
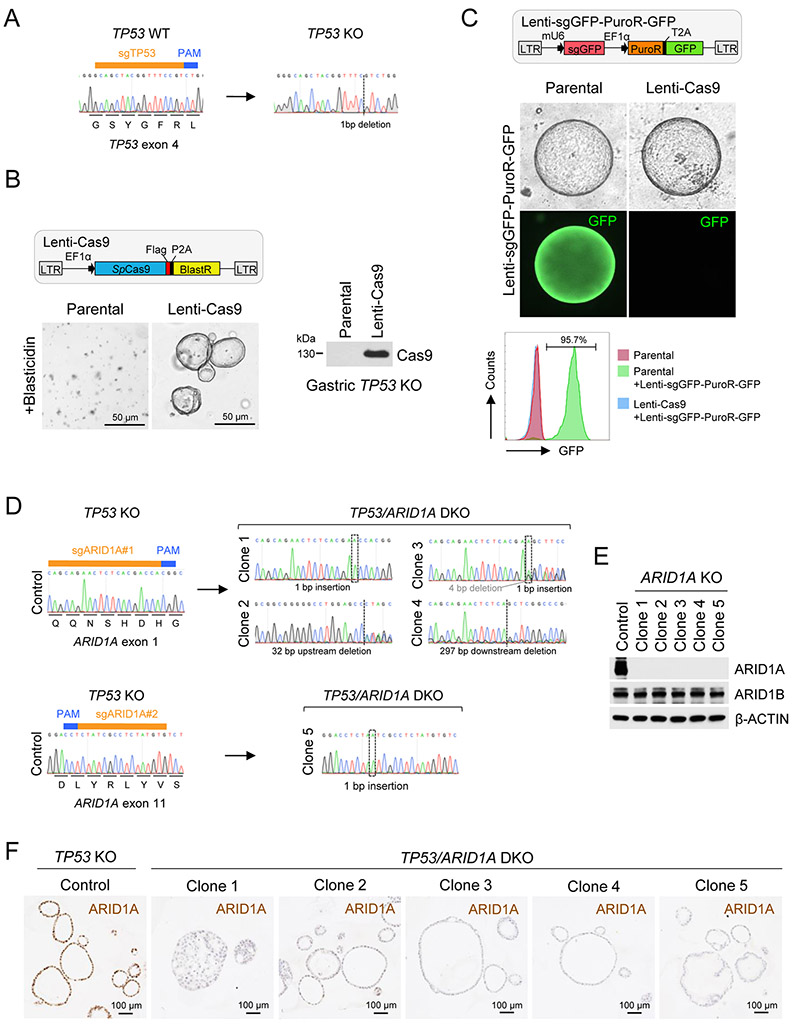
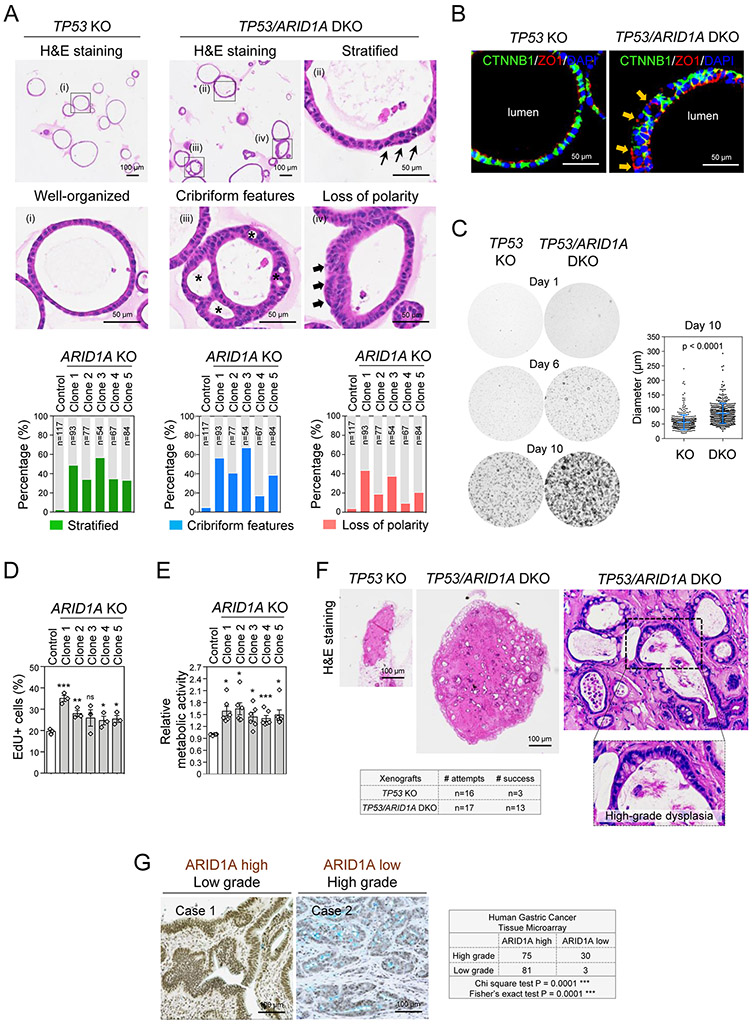
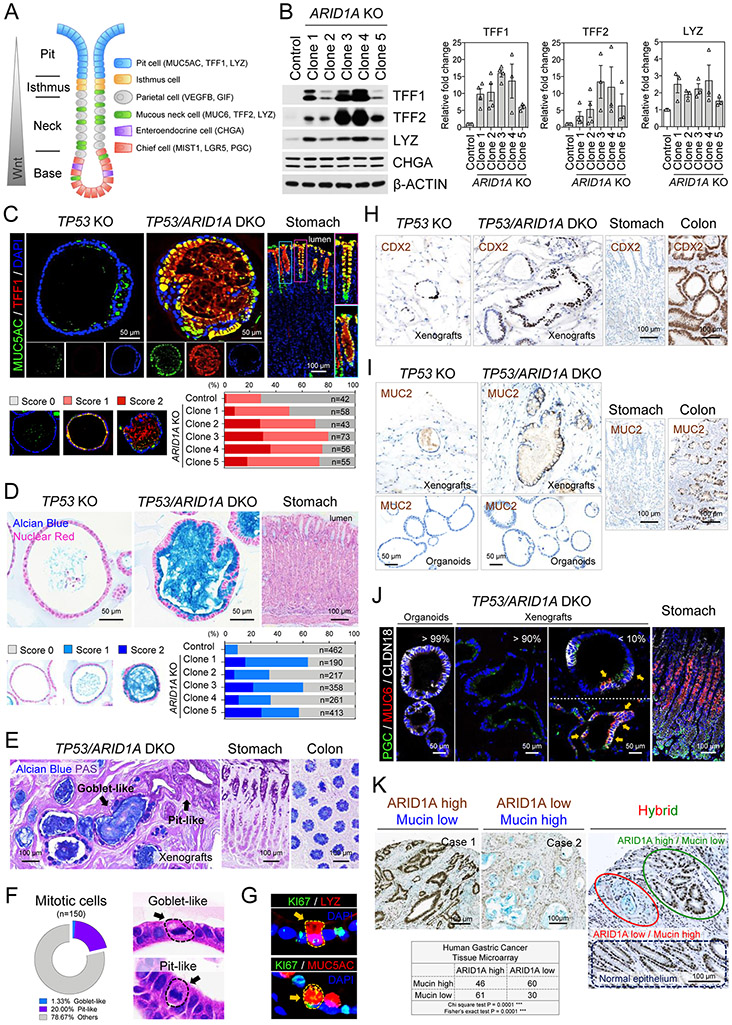
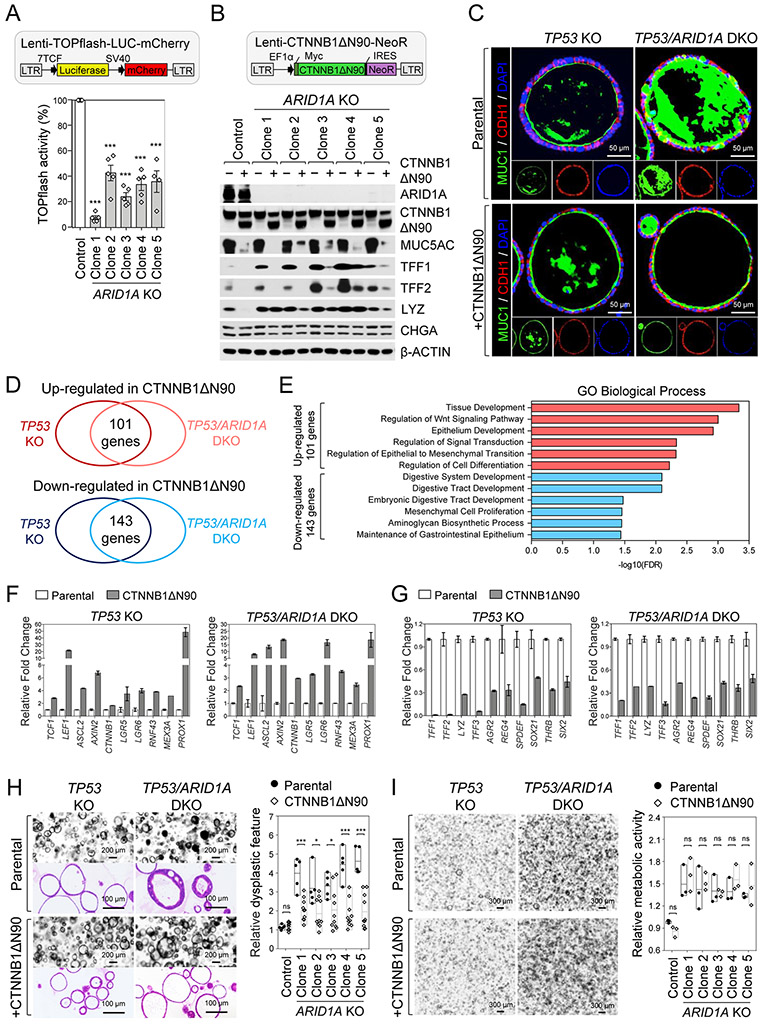
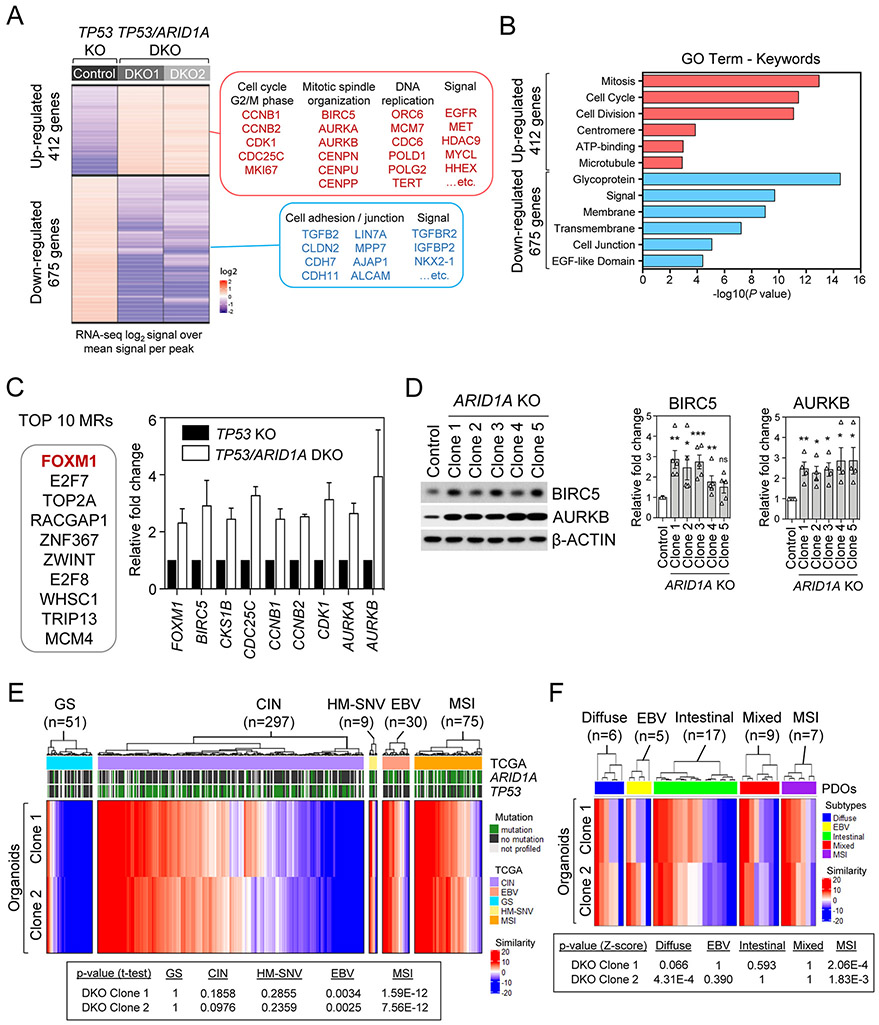
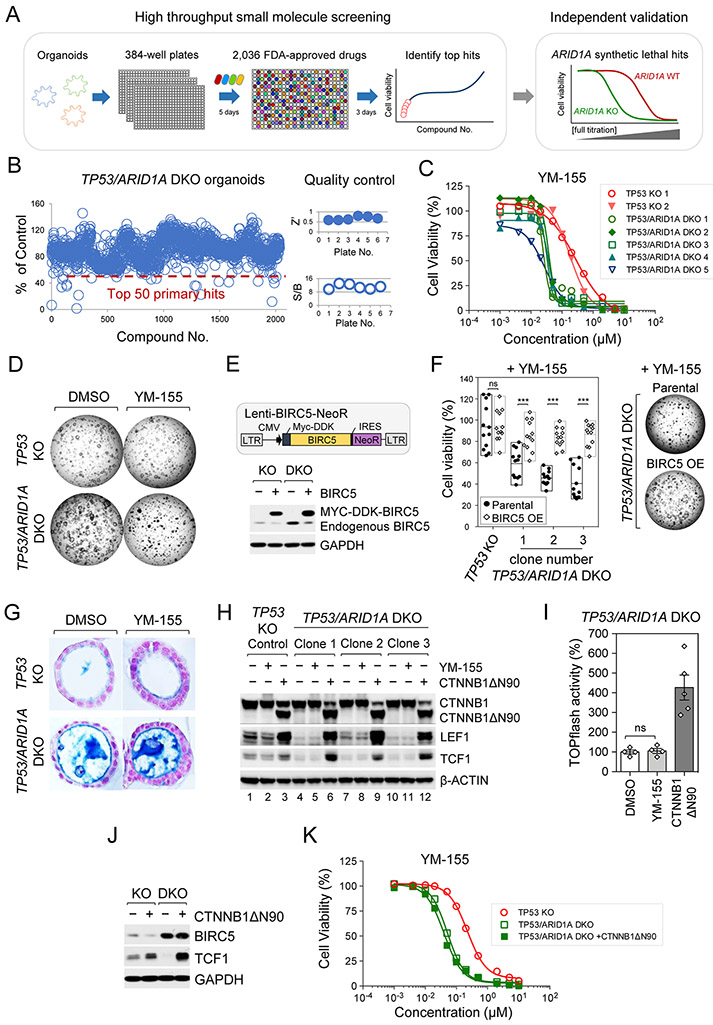
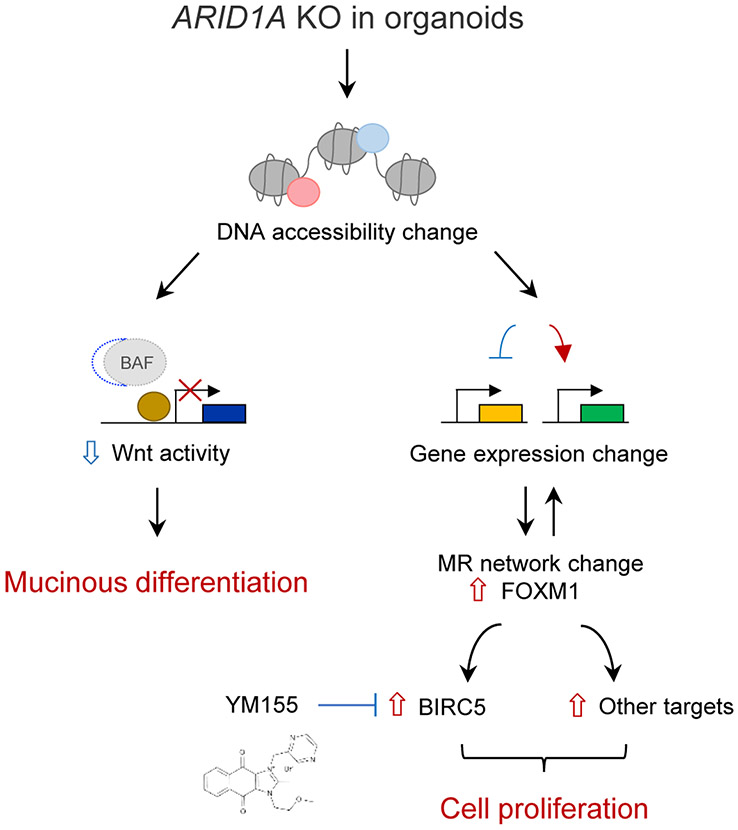
Mutations in ARID1A rank among the most common molecular aberrations in human cancer. However, oncogenic consequences of ARID1A mutation in human cells remain poorly defined due to lack of forward genetic models. Here, CRISPR/Cas9-mediated ARID1A knockout (KO) in primary TP53-/- human gastric organoids induced morphologic dysplasia, tumorigenicity, and mucinous differentiation. Genetic WNT/β-catenin activation rescued mucinous differentiation, but not hyperproliferation, suggesting alternative pathways of ARID1A KO-mediated transformation. ARID1A mutation induced transcriptional regulatory modules characteristic of microsatellite instability and Epstein-Barr virus-associated subtype human gastric cancer, including FOXM1-associated mitotic genes and BIRC5/survivin. Convergently, high-throughput compound screening indicated selective vulnerability of ARID1A-deficient organoids to inhibition of BIRC5/survivin, functionally implicating this pathway as an essential mediator of ARID1A KO-dependent early-stage gastric tumorigenesis. Overall, we define distinct pathways downstream of oncogenic ARID1A mutation, with nonessential WNT-inhibited mucinous differentiation in parallel with essential transcriptional FOXM1/BIRC5-stimulated proliferation, illustrating the general utility of organoid-based forward genetic cancer analysis in human cells. SIGNIFICANCE: We establish the first human forward genetic modeling of a commonly mutated tumor suppressor gene, ARID1A. Our study integrates diverse modalities including CRISPR/Cas9 genome editing, organoid culture, systems biology, and small-molecule screening to derive novel insights into early transformation mechanisms of ARID1A-deficient gastric cancers.See related commentary by Zafra and Dow, p. 1327.This article is highlighted in the In This Issue feature, p. 1307.
©2021 American Association for Cancer Research.
Conflict of interest statement
Conflict of Interest Statement
The authors declare no potential conflicts of interest
Figures
Comment in
-
Revealing ARID1A Function in Gastric Cancer from the Bottom Up.Cancer Discov. 2021 Jun;11(6):1327-1329. doi: 10.1158/2159-8290.CD-21-0271. Cancer Discov. 2021. PMID: 34078662
References
Publication types
MeSH terms
Substances
Grants and funding
- K00 CA212433/CA/NCI NIH HHS/United States
- U54 CA224081/CA/NCI NIH HHS/United States
- U01 CA199241/CA/NCI NIH HHS/United States
- U01 CA217875/CA/NCI NIH HHS/United States
- S10 OD012351/OD/NIH HHS/United States
- K99 GM134154/GM/NIGMS NIH HHS/United States
- R35 CA197745/CA/NCI NIH HHS/United States
- R00 GM134154/GM/NIGMS NIH HHS/United States
- U01 DK085527/DK/NIDDK NIH HHS/United States
- R01 CA163915/CA/NCI NIH HHS/United States
- U01 CA217858/CA/NCI NIH HHS/United States
- U01 CA217851/CA/NCI NIH HHS/United States
- U19 AI116484/AI/NIAID NIH HHS/United States
- S10 OD021764/OD/NIH HHS/United States
- U01 CA217883/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
