

Bacteria and Metabolic Potential in Karst Caves Revealed by Intensive Bacterial Cultivation and Genome Assembly
- PMID: 33452024
- PMCID: PMC8105019
- DOI: 10.1128/AEM.02440-20
Bacteria and Metabolic Potential in Karst Caves Revealed by Intensive Bacterial Cultivation and Genome Assembly
Erratum in
-
Correction for Zhu et al., "Bacteria and Metabolic Potential in Karst Caves Revealed by Intensive Bacterial Cultivation and Genome Assembly".Appl Environ Microbiol. 2021 May 26;87(12):e0057721. doi: 10.1128/AEM.00577-21. Epub 2021 May 26. Appl Environ Microbiol. 2021. PMID: 34037461 Free PMC article. No abstract available.
Abstract
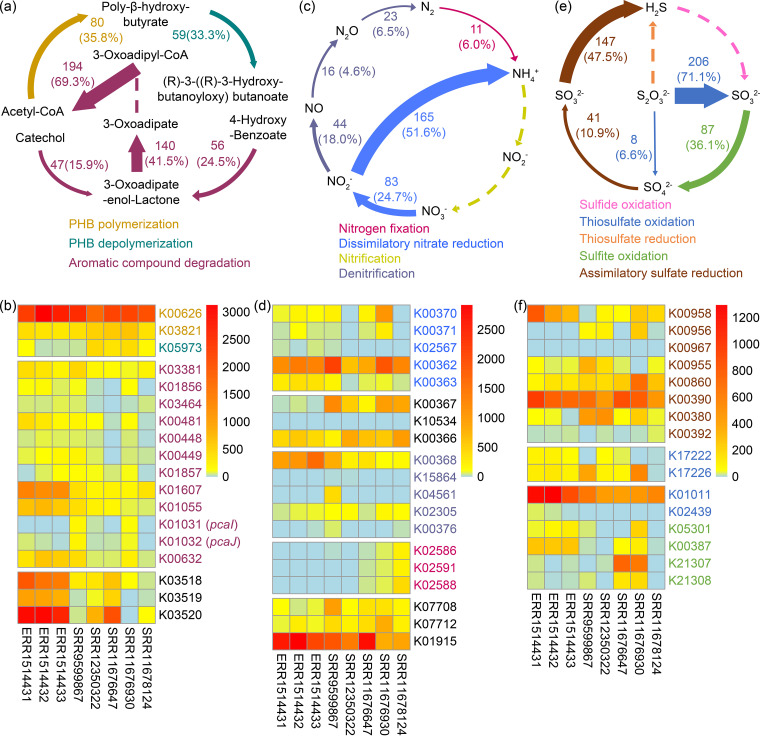
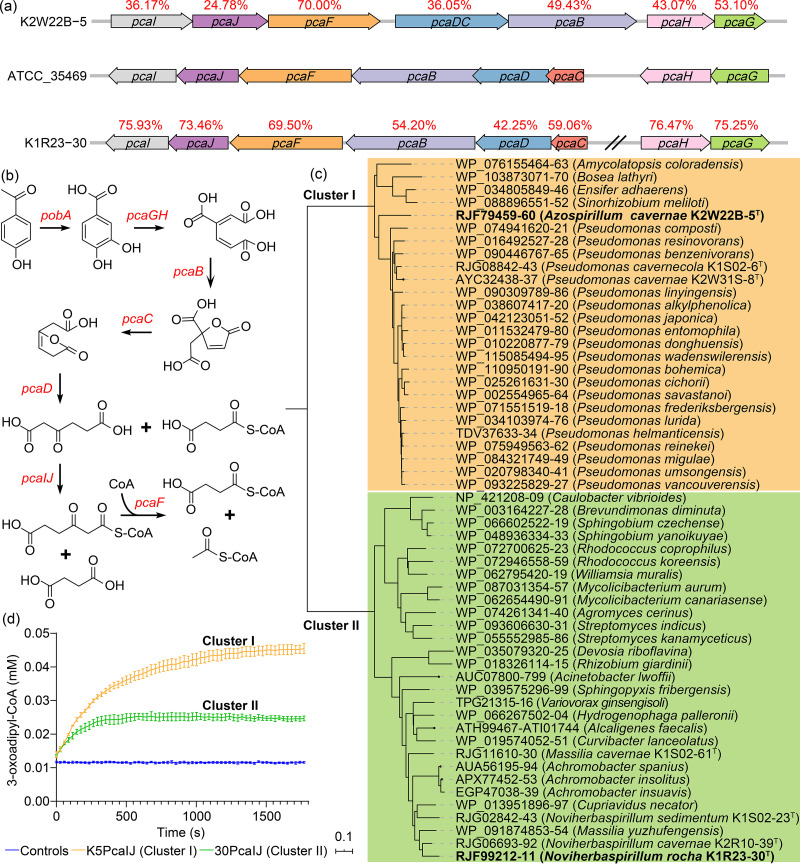
Karst caves are widely distributed subsurface systems, and the microbiomes therein are proposed to be the driving force for cave evolution and biogeochemical cycling. In past years, culture-independent studies on the microbiomes of cave systems have been conducted, yet intensive microbial cultivation is still needed to validate the sequence-derived hypothesis and to disclose the microbial functions in cave ecosystems. In this study, the microbiomes of two karst caves in Guizhou Province in southwest China were examined. A total of 3,562 bacterial strains were cultivated from rock, water, and sediment samples, and 329 species (including 14 newly described species) of 102 genera were found. We created a cave bacterial genome collection of 218 bacterial genomes from a karst cave microbiome through the extraction of 204 database-derived genomes and de novo sequencing of 14 new bacterial genomes. The cultivated genome collection obtained in this study and the metagenome data from previous studies were used to investigate the bacterial metabolism and potential involvement in the carbon, nitrogen, and sulfur biogeochemical cycles in the cave ecosystem. New N2-fixing Azospirillum and alkane-oxidizing Oleomonas species were documented in the karst cave microbiome. Two pcaIJ clusters of the β-ketoadipate pathway that were abundant in both the cultivated microbiomes and the metagenomic data were identified, and their representatives from the cultivated bacterial genomes were functionally demonstrated. This large-scale cultivation of a cave microbiome represents the most intensive collection of cave bacterial resources to date and provides valuable information and diverse microbial resources for future cave biogeochemical research.IMPORTANCE Karst caves are oligotrophic environments that are dark and humid and have a relatively stable annual temperature. The diversity of bacteria and their metabolisms are crucial for understanding the biogeochemical cycling in cave ecosystems. We integrated large-scale bacterial cultivation with metagenomic data mining to explore the compositions and metabolisms of the microbiomes in two karst cave systems. Our results reveal the presence of a highly diversified cave bacterial community, and 14 new bacterial species were described and their genomes sequenced. In this study, we obtained the most intensive collection of cultivated microbial resources from karst caves to date and predicted the various important routes for the biogeochemical cycling of elements in cave ecosystems.
Keywords: 3-oxoadipate-CoA transferases; Azospirillum; Oleomonas; bacterial cultivation; biogeochemical cycling; karst cave microbiome.
Copyright © 2021 American Society for Microbiology.
Figures
Similar articles
-
Dominant bacterial phyla in caves and their predicted functional roles in C and N cycle.BMC Microbiol. 2017 Apr 11;17(1):90. doi: 10.1186/s12866-017-1002-x. BMC Microbiol. 2017. PMID: 28399822 Free PMC article.
-
Co-occurrence pattern and function prediction of bacterial community in Karst cave.BMC Microbiol. 2020 May 29;20(1):137. doi: 10.1186/s12866-020-01806-7. BMC Microbiol. 2020. PMID: 32471344 Free PMC article.
-
Shotgun metagenomic sequencing from Manao-Pee cave, Thailand, reveals insight into the microbial community structure and its metabolic potential.BMC Microbiol. 2019 Jun 27;19(1):144. doi: 10.1186/s12866-019-1521-8. BMC Microbiol. 2019. PMID: 31248378 Free PMC article.
-
Microbial roles in cave biogeochemical cycling.Front Microbiol. 2022 Sep 28;13:950005. doi: 10.3389/fmicb.2022.950005. eCollection 2022. Front Microbiol. 2022. PMID: 36246268 Free PMC article. Review.
-
Microbial ecology of tourist Paleolithic caves.Sci Total Environ. 2022 Apr 10;816:151492. doi: 10.1016/j.scitotenv.2021.151492. Epub 2021 Nov 15. Sci Total Environ. 2022. PMID: 34793801 Review.
Cited by
-
The Progression in Developing Genomic Resources for Crop Improvement.Life (Basel). 2023 Jul 31;13(8):1668. doi: 10.3390/life13081668. Life (Basel). 2023. PMID: 37629524 Free PMC article. Review.
-
Expanding the bioanalytical application of β-hydroxybutyrate binding proteins through characterization of their metabolite interactions and site-directed mutagenesis.Protein Sci. 2025 May;34(5):e70129. doi: 10.1002/pro.70129. Protein Sci. 2025. PMID: 40260974
-
The Saint-Leonard Urban Glaciotectonic Cave Harbors Rich and Diverse Planktonic and Sedimentary Microbial Communities.Microorganisms. 2024 Aug 29;12(9):1791. doi: 10.3390/microorganisms12091791. Microorganisms. 2024. PMID: 39338466 Free PMC article.
-
Microbial Interactions Drive Distinct Taxonomic and Potential Metabolic Responses to Habitats in Karst Cave Ecosystem.Microbiol Spectr. 2021 Oct 31;9(2):e0115221. doi: 10.1128/Spectrum.01152-21. Epub 2021 Sep 8. Microbiol Spectr. 2021. PMID: 34494852 Free PMC article.
-
Carbonatogenic Bacteria in the Maros-Pangkep Karst: Protectors or Threat to Prehistoric Paintings?J Microbiol Biotechnol. 2025 Feb 20;35:e2410019. doi: 10.4014/jmb.2410.10019. J Microbiol Biotechnol. 2025. PMID: 40016134 Free PMC article.
References
-
- Gabriel CR, Northup DE. 2013. Microbial ecology: caves as an extreme habitat, p 85–108. In Cheeptham N (ed), Cave microbiomes: a novel resource for drug discovery. Springer, New York, NY.
-
- Kuzmina LY, Galimzianova NF, Abdullin SR, Ryabova AS. 2012. Microbiota of the Kinderlinskaya cave (South Urals, Russia). Microbiology 81:251–258. doi:10.1134/S0026261712010109. - DOI
-
- Barton HA, Jurado V. 2007. What's up down there? Microbial diversity in caves, p 132–138. In Schaechter M (ed), Microbe. ASM Press, Washington, DC.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
