

Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications
- PMID: 33452396
- PMCID: PMC7810997
- DOI: 10.1038/s41598-021-81093-y
Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications
Erratum in
-
Author Correction: Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications.Sci Rep. 2021 May 10;11(1):10340. doi: 10.1038/s41598-021-89275-4. Sci Rep. 2021. PMID: 33972629 Free PMC article. No abstract available.
Abstract
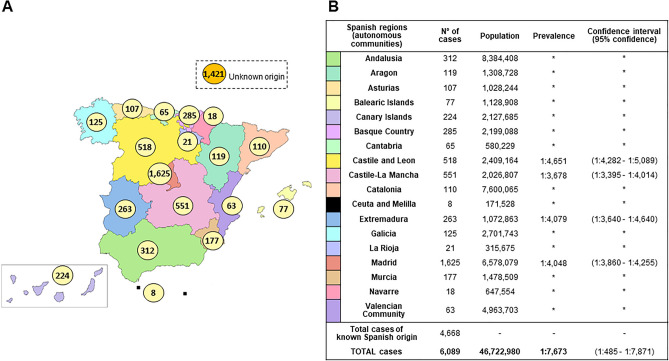
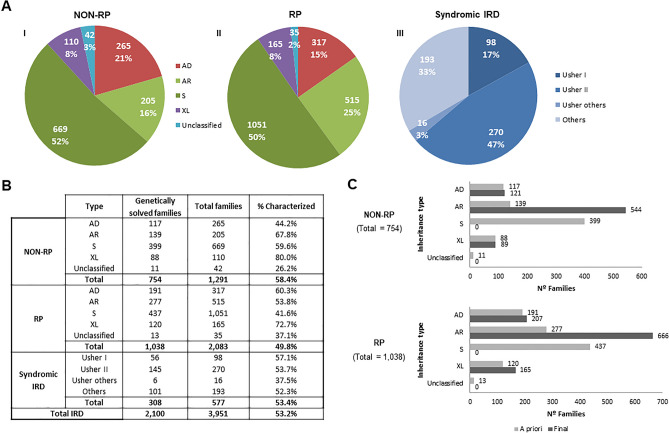
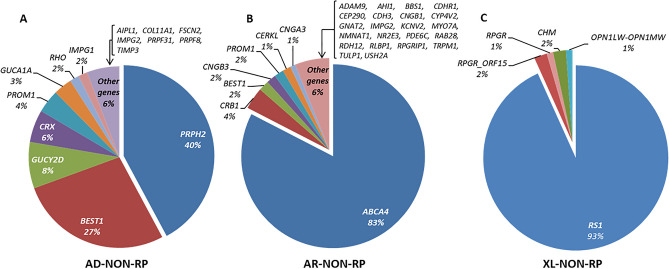
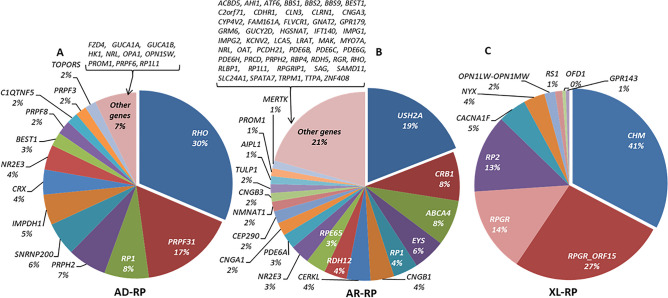
Inherited retinal diseases (IRDs), defined by dysfunction or progressive loss of photoreceptors, are disorders characterized by elevated heterogeneity, both at the clinical and genetic levels. Our main goal was to address the genetic landscape of IRD in the largest cohort of Spanish patients reported to date. A retrospective hospital-based cross-sectional study was carried out on 6089 IRD affected individuals (from 4403 unrelated families), referred for genetic testing from all the Spanish autonomous communities. Clinical, demographic and familiar data were collected from each patient, including family pedigree, age of appearance of visual symptoms, presence of any systemic findings and geographical origin. Genetic studies were performed to the 3951 families with available DNA using different molecular techniques. Overall, 53.2% (2100/3951) of the studied families were genetically characterized, and 1549 different likely causative variants in 142 genes were identified. The most common phenotype encountered is retinitis pigmentosa (RP) (55.6% of families, 2447/4403). The most recurrently mutated genes were PRPH2, ABCA4 and RS1 in autosomal dominant (AD), autosomal recessive (AR) and X-linked (XL) NON-RP cases, respectively; RHO, USH2A and RPGR in AD, AR and XL for non-syndromic RP; and USH2A and MYO7A in syndromic IRD. Pathogenic variants c.3386G > T (p.Arg1129Leu) in ABCA4 and c.2276G > T (p.Cys759Phe) in USH2A were the most frequent variants identified. Our study provides the general landscape for IRD in Spain, reporting the largest cohort ever presented. Our results have important implications for genetic diagnosis, counselling and new therapeutic strategies to both the Spanish population and other related populations.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Vaidya P, Vaidya A. Retinitis pigmentosa: Disease encumbrance in the eurozone. Int. J. Ophthalmol. Clin. Res. 2015;2:030. doi: 10.23937/2378-346x/1410030. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
