

Computational methods for the prediction of chromatin interaction and organization using sequence and epigenomic profiles
- PMID: 33454752
- PMCID: PMC8424394
- DOI: 10.1093/bib/bbaa405
Computational methods for the prediction of chromatin interaction and organization using sequence and epigenomic profiles
Abstract
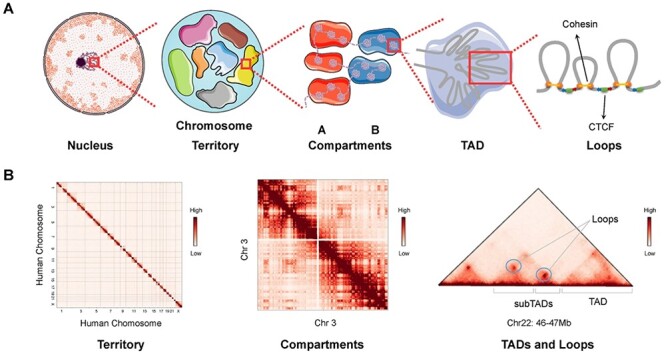
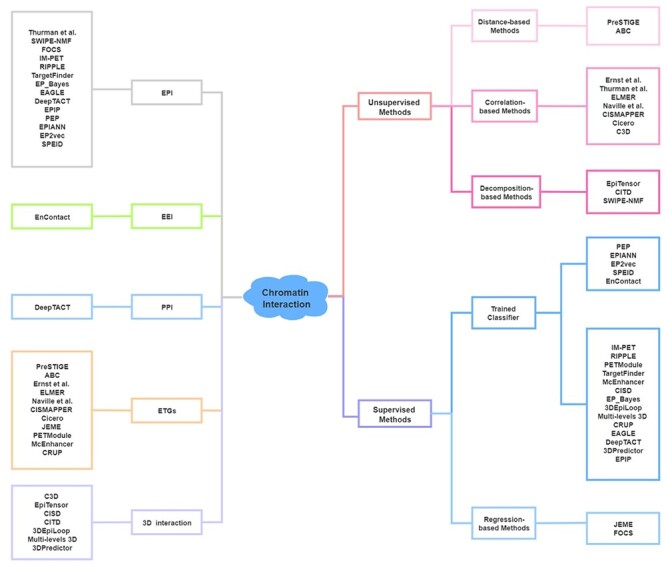
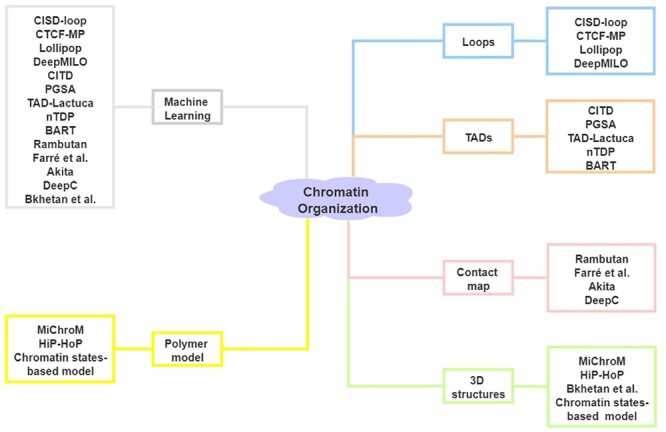
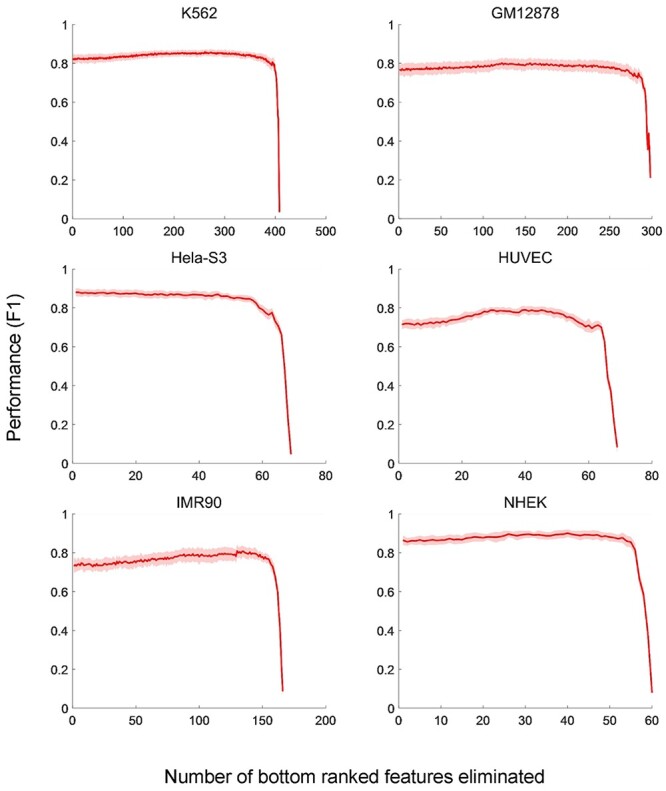
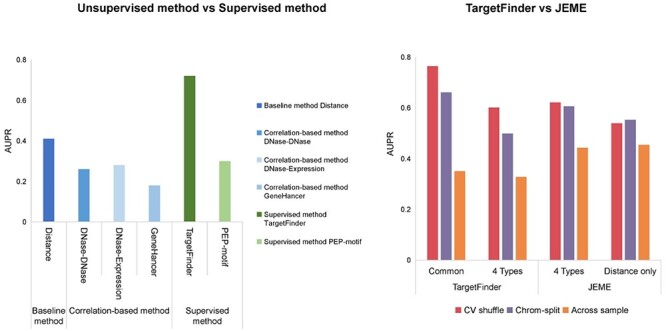
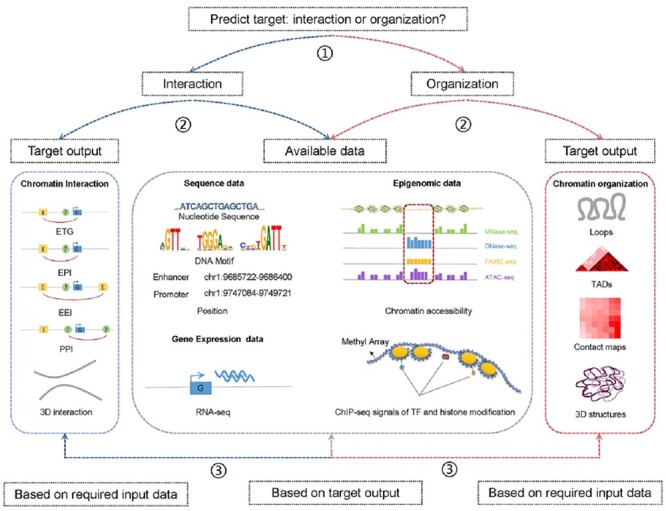
The exploration of three-dimensional chromatin interaction and organization provides insight into mechanisms underlying gene regulation, cell differentiation and disease development. Advances in chromosome conformation capture technologies, such as high-throughput chromosome conformation capture (Hi-C) and chromatin interaction analysis by paired-end tag (ChIA-PET), have enabled the exploration of chromatin interaction and organization. However, high-resolution Hi-C and ChIA-PET data are only available for a limited number of cell lines, and their acquisition is costly, time consuming, laborious and affected by theoretical limitations. Increasing evidence shows that DNA sequence and epigenomic features are informative predictors of regulatory interaction and chromatin architecture. Based on these features, numerous computational methods have been developed for the prediction of chromatin interaction and organization, whereas they are not extensively applied in biomedical study. A systematical study to summarize and evaluate such methods is still needed to facilitate their application. Here, we summarize 48 computational methods for the prediction of chromatin interaction and organization using sequence and epigenomic profiles, categorize them and compare their performance. Besides, we provide a comprehensive guideline for the selection of suitable methods to predict chromatin interaction and organization based on available data and biological question of interest.
Keywords: DNA sequence and epigenomic features; gene regulation; methods evaluation; three-dimensional genome organization.
© The Author(s) 2021. Published by Oxford University Press.
Figures
References
-
- Dekker J, Rippe K, Dekker M, et al. . Capturing chromosome conformation. Science 2002;295:1306–11. - PubMed
-
- Simonis M, Klous P, Splinter E, et al. . Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet 2006;38:1348–54. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
