

Advances in quantitative high-throughput phosphoproteomics with sample multiplexing
- PMID: 33455035
- PMCID: PMC8209658
- DOI: 10.1002/pmic.202000140
Advances in quantitative high-throughput phosphoproteomics with sample multiplexing
Abstract
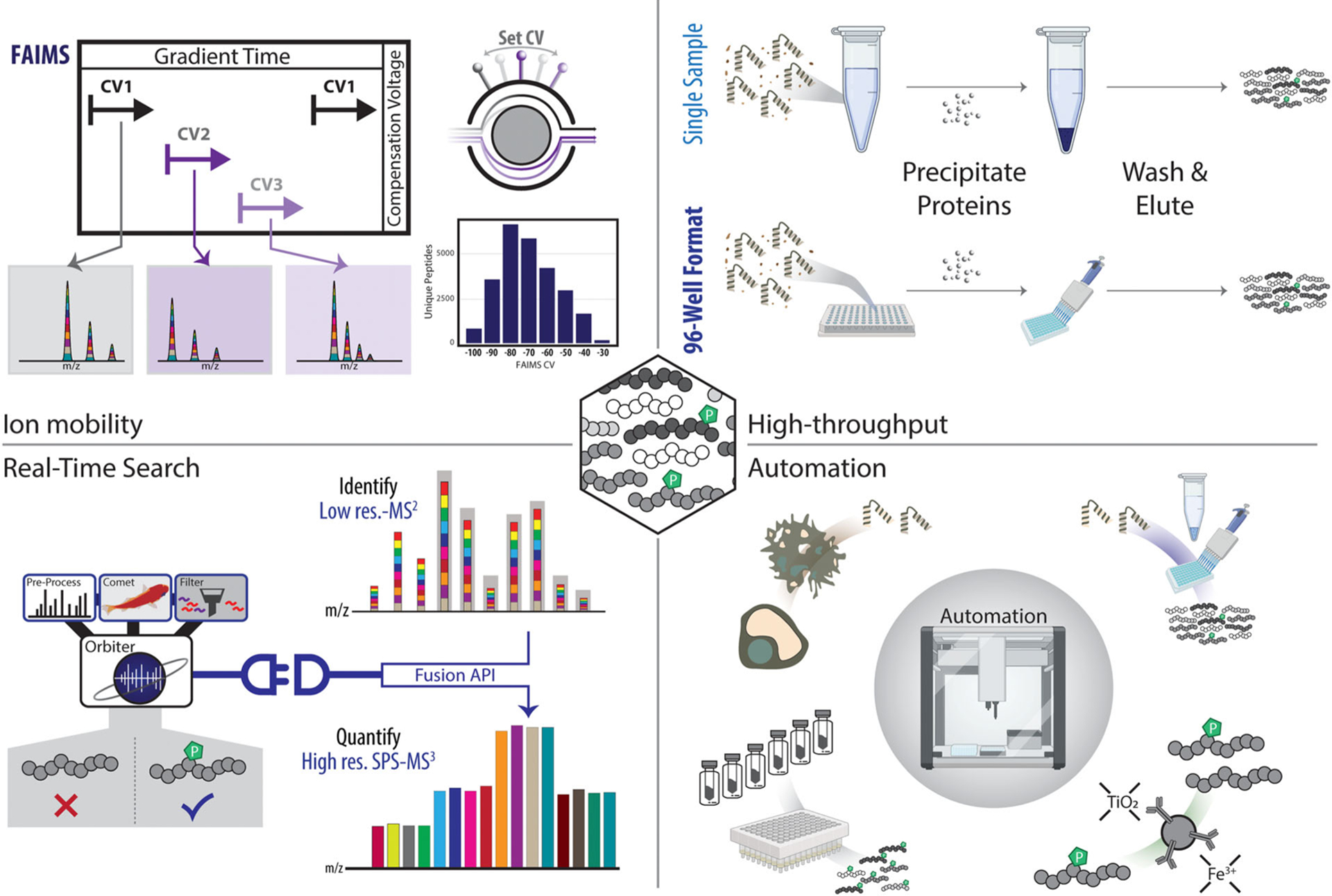
Eukaryotic protein phosphorylation modulates nearly every major biological process. Phosphorylation regulates protein activity, mediates cellular signal transduction, and manipulates cellular structure. Consequently, the dysregulation of kinase and phosphatase pathways has been linked to a multitude of diseases. Mass spectrometry-based proteomic techniques are increasingly used for the global interrogation of perturbations in phosphorylation-based cellular signaling. Strategies for studying phosphoproteomes require high-specificity enrichment, sensitive detection, and accurate localization of phosphorylation sites with advanced LC-MS/MS techniques and downstream informatics. Sample multiplexing with isobaric tags has also been integral to recent advancements in throughput and sensitivity for phosphoproteomic studies. Each of these facets of phosphoproteomics analysis present distinct challenges and thus opportunities for improvement and innovation. Here, we review current methodologies, explore persistent challenges, and discuss the outlook for isobaric tag-based quantitative phosphoproteomic analysis.
Keywords: automation; high-throughput; tandem-mass tag.
© 2021 Wiley-VCH GmbH.
Conflict of interest statement
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Figures



References
-
- Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nar-done J, Lee K, Reeves C, Li Y, Hu Y, Tan Z, Stokes M, Sullivan L, Mitchell J, Wetzel R, Macneill J, Ren JM, Yuan J, Bakalarski CE, Villen J, Kornhauser JM, Smith B, Li D, Zhou X, Gygi SP, Gu TL, Polakiewicz RD, Rush J, & Comb MJ (2007). Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell, 131, 1190–1203; - PubMed
- Zanivan S, Meves A, Behrendt K, Schoof EM, Neilson LJ, Cox J, Tang HR, Kalna G, van Ree JH, van Deursen JM, Trempus CS, Machesky LM, Linding R, Wickstrom SA, Fassler R, & Mann M (2013). In vivo SILAC-based proteomics reveals phosphoproteome changes during mouse skin carcinogenesis. Cell Reports, 3, 552–66. - PubMed
-
- Danielsson A, Ost A, Nystrom FH, & Stralfors P (2005). Cttenuation of insulin-stimulated insulin receptor substrate-1 serine 307 phosphorylation in insulin resistance of type 2 diabetes. Journal of Biological Chemistry, 280, 34389–34392. - PubMed
-
- Eidenmuller J, Fath T, Maas T, Pool M, Sontag E, & Brandt R (2001). Phosphorylation-mimicking glutamate clusters in the proline-rich region are sufficient to simulate the functional deficiencies of hyperphosphorylated tau protein. Biochemical Journal, 357, 759–759; - PMC - PubMed
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, & Binder LI (1986). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. PNAS, 83, 4913–4917. - PMC - PubMed
-
- Manning G, Plowman GD, Hunter T, & Sudarsanam S (2002). Evolution of protein kinase signaling from yeast to man. Trends in Biochemical Sciences, 27, 514–520. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
