

Implementation of adaptive integration method for free energy calculations in molecular systems
- PMID: 33457645
- PMCID: PMC7808261
- DOI: 10.7717/peerj-cs.264
Implementation of adaptive integration method for free energy calculations in molecular systems
Abstract
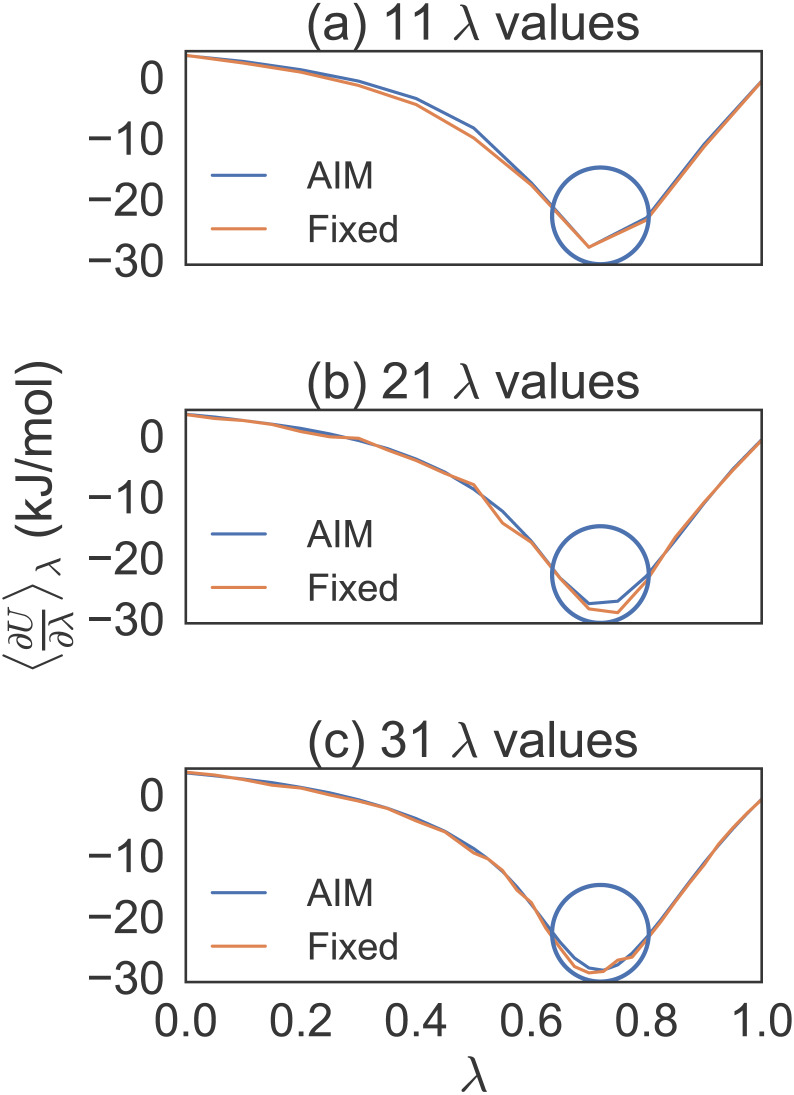
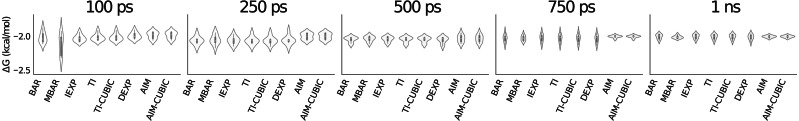
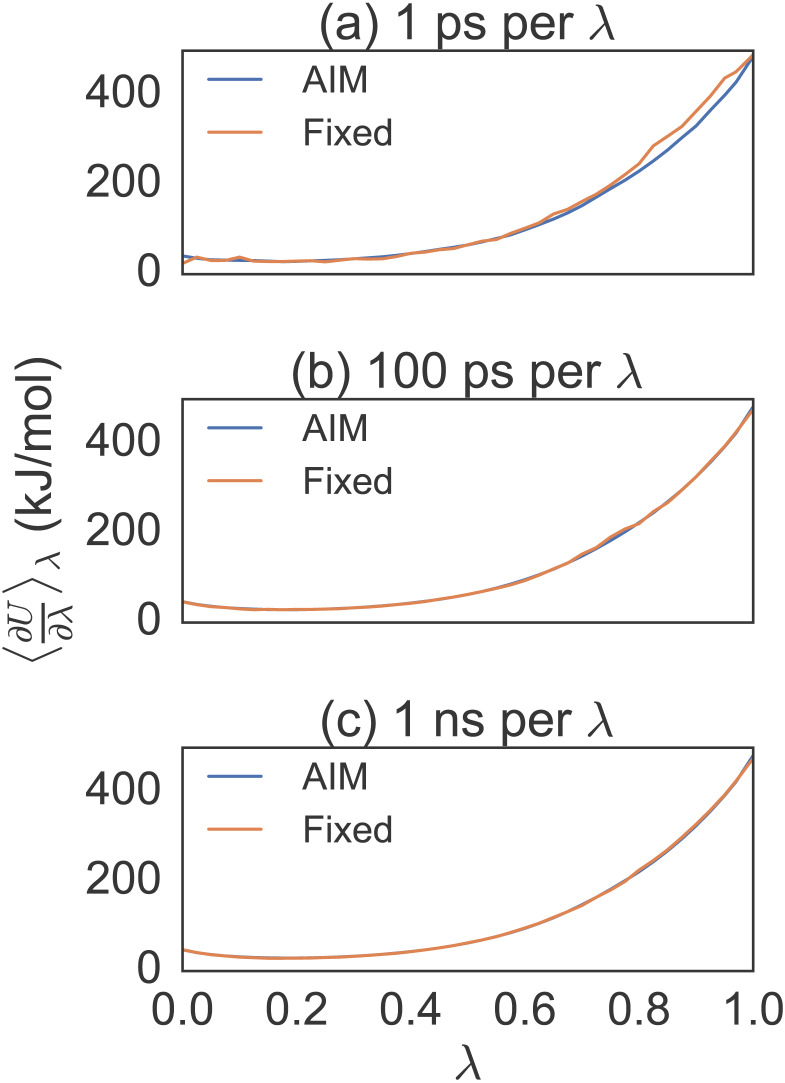
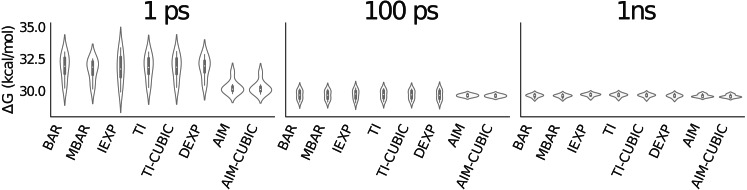
Estimating free energy differences by computer simulation is useful for a wide variety of applications such as virtual screening for drug design and for understanding how amino acid mutations modify protein interactions. However, calculating free energy differences remains challenging and often requires extensive trial and error and very long simulation times in order to achieve converged results. Here, we present an implementation of the adaptive integration method (AIM). We tested our implementation on two molecular systems and compared results from AIM to those from a suite of other methods. The model systems tested here include calculating the solvation free energy of methane, and the free energy of mutating the peptide GAG to GVG. We show that AIM is more efficient than other tested methods for these systems, that is, AIM results converge to a higher level of accuracy and precision for a given simulation time.
Keywords: Adaptive integration; Biomolecule; Computational Biology; Free energy; Monte Carlo; Protein; Scientific Computing and Simulation; Solvation.
Conflict of interest statement
Competing Interests The authors declare there are no competing interests.
Figures
Similar articles
-
Increasing the Efficiency of Free Energy Calculations Using Parallel Tempering and Histogram Reweighting.J Chem Theory Comput. 2006 Jul;2(4):939-46. doi: 10.1021/ct050207o. J Chem Theory Comput. 2006. PMID: 26633053
-
Accurate estimation of the density of states from Monte Carlo transition probability data.J Chem Phys. 2006 Oct 14;125(14):144905. doi: 10.1063/1.2358345. J Chem Phys. 2006. PMID: 17042648
-
Advancing Drug Discovery through Enhanced Free Energy Calculations.Acc Chem Res. 2017 Jul 18;50(7):1625-1632. doi: 10.1021/acs.accounts.7b00083. Epub 2017 Jul 5. Acc Chem Res. 2017. PMID: 28677954
-
Development and test of highly accurate endpoint free energy methods. 1: Evaluation of ABCG2 charge model on solvation free energy prediction and optimization of atom radii suitable for more accurate solvation free energy prediction by the PBSA method.J Comput Chem. 2023 May 30;44(14):1334-1346. doi: 10.1002/jcc.27089. Epub 2023 Feb 21. J Comput Chem. 2023. PMID: 36807356 Review.
-
Comparison of free energy methods for molecular systems.J Chem Phys. 2006 Nov 14;125(18):184114. doi: 10.1063/1.2378907. J Chem Phys. 2006. PMID: 17115745 Review.
References
-
- Abraham M, Van der Spoel D, Lindahl E, Hess B. GROMACS user manual version 5.1.4. http//:www.gromacs.org 2016
-
- Bennett CH. Efficient estimation of free energy differences from Monte Carlo Data. Journal of Computational Physics. 1976;22:245–268. doi: 10.1016/0021-9991(76)90078-4. - DOI
-
- Berendsen HJ, Van der Spoel D, Van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Computer Physics Communications. 1995;91(1–3):43–56. doi: 10.1016/0010-4655(95)00042-E. - DOI
-
- Bitetti-Putzer R, Yang W, Karplus M. Generalized ensembles serve to improve the convergence of free energy simulations. Chemical Physics Letters. 2003;377(5–6):633–641. doi: 10.1016/S0009-2614(03)01057-1. - DOI
Grants and funding
LinkOut - more resources
Full Text Sources