

The rapidly evolving view of lysosomal storage diseases
- PMID: 33459519
- PMCID: PMC7863408
- DOI: 10.15252/emmm.202012836
The rapidly evolving view of lysosomal storage diseases
Abstract
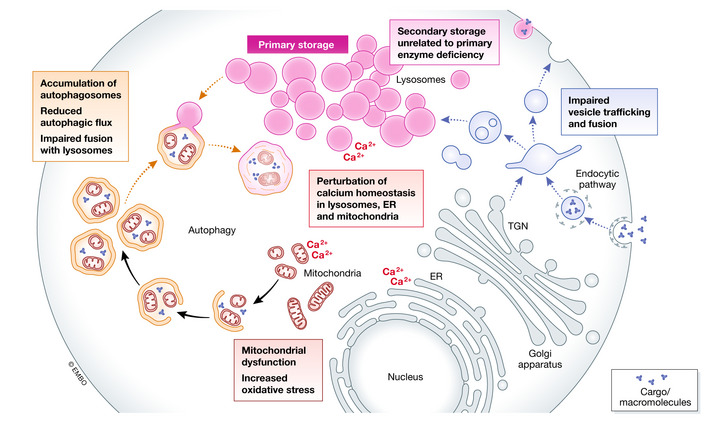
Lysosomal storage diseases are a group of metabolic disorders caused by deficiencies of several components of lysosomal function. Most commonly affected are lysosomal hydrolases, which are involved in the breakdown and recycling of a variety of complex molecules and cellular structures. The understanding of lysosomal biology has progressively improved over time. Lysosomes are no longer viewed as organelles exclusively involved in catabolic pathways, but rather as highly dynamic elements of the autophagic-lysosomal pathway, involved in multiple cellular functions, including signaling, and able to adapt to environmental stimuli. This refined vision of lysosomes has substantially impacted on our understanding of the pathophysiology of lysosomal disorders. It is now clear that substrate accumulation triggers complex pathogenetic cascades that are responsible for disease pathology, such as aberrant vesicle trafficking, impairment of autophagy, dysregulation of signaling pathways, abnormalities of calcium homeostasis, and mitochondrial dysfunction. Novel technologies, in most cases based on high-throughput approaches, have significantly contributed to the characterization of lysosomal biology or lysosomal dysfunction and have the potential to facilitate diagnostic processes, and to enable the identification of new therapeutic targets.
Keywords: autophagy; lysosomal biology; lysosomal storage diseases; lysosomes.
© 2021 The Authors. Published under the terms of the CC BY 4.0 license.
Conflict of interest statement
A. Ballabio is co‐founder of CASMA Therapeutics and of Next Generation Diagnostics (NGD).
Figures

References
-
- A Study to Assess the Long‐term Safety and Efficacy of ATB200/AT2221 in Adult Subjects with LOPD (2019) ClinicalTrials.Gov NCT04138277 (https://clinicaltrials.gov/ct2/show/NCT04138277) [DATASET]
-
- Aharon‐Peretz J, Rosenbaum H, Gershoni‐Baruch R (2004) Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med 351: 1972–1977 - PubMed
-
- Anderson S (2018) Newborn screening for lysosomal storage disorders. J Pediatr Health Care 32: 285–294 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
