

Regulatory roles of Escherichia coli 5' UTR and ORF-internal RNAs detected by 3' end mapping
- PMID: 33460557
- PMCID: PMC7815308
- DOI: 10.7554/eLife.62438
Regulatory roles of Escherichia coli 5' UTR and ORF-internal RNAs detected by 3' end mapping
Erratum in
-
Correction: Regulatory roles of Escherichia coli 5' UTR and ORF-internal RNAs detected by 3' end mapping.Elife. 2021 Apr 13;10:e69260. doi: 10.7554/eLife.69260. Elife. 2021. PMID: 33847262 Free PMC article.
Abstract
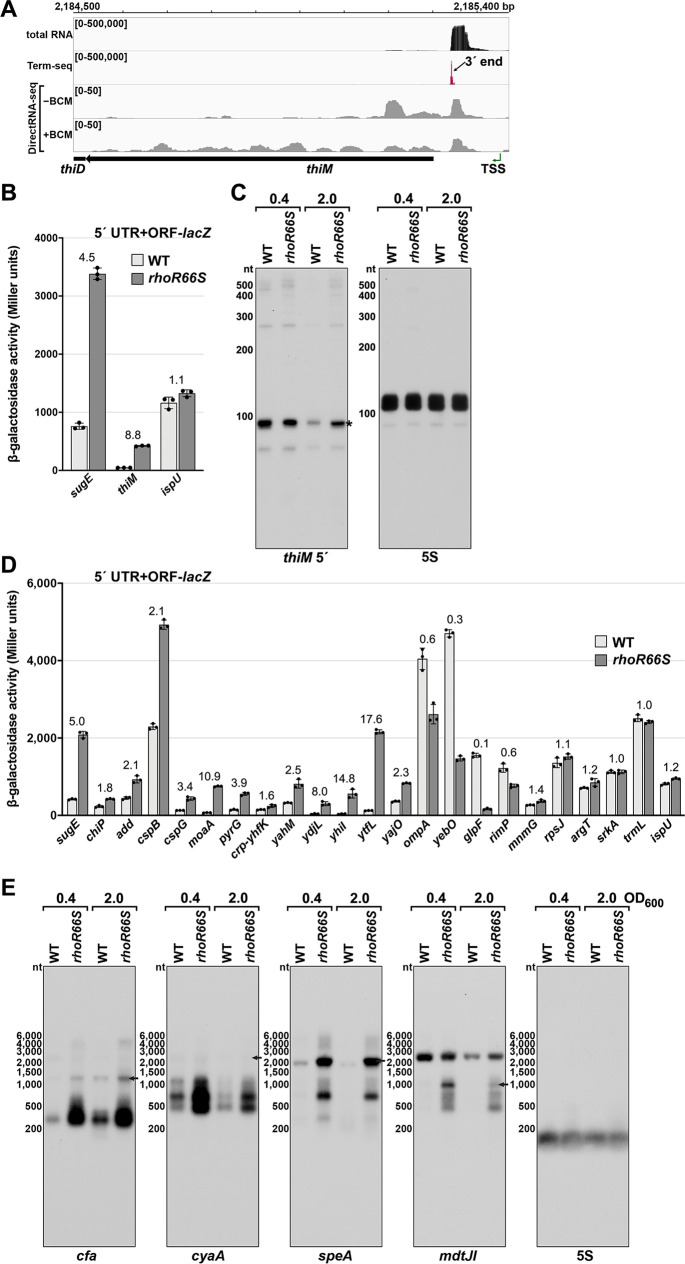
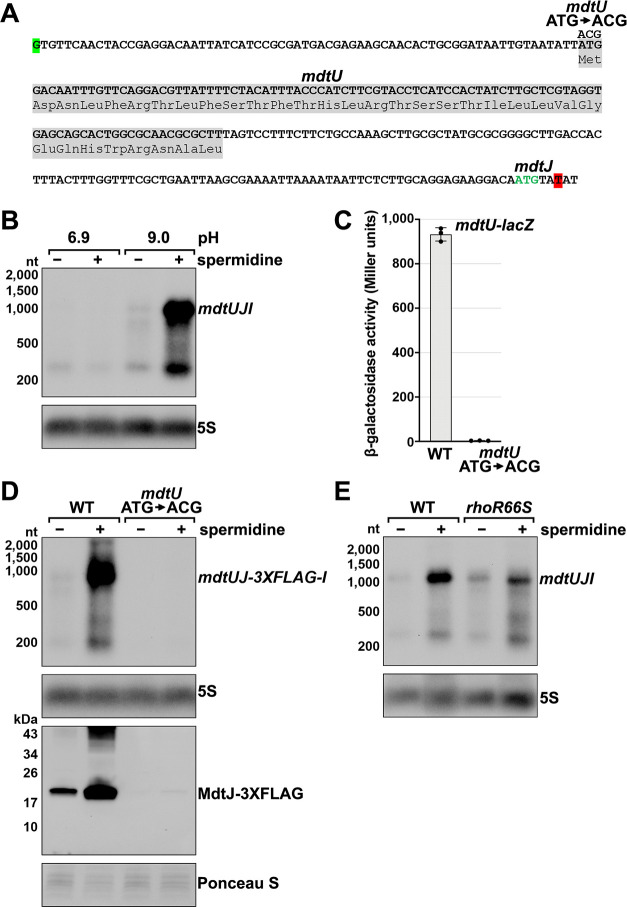
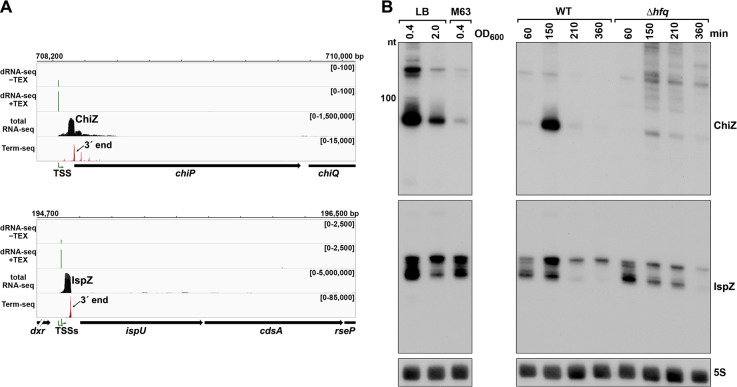
Many bacterial genes are regulated by RNA elements in their 5´ untranslated regions (UTRs). However, the full complement of these elements is not known even in the model bacterium Escherichia coli. Using complementary RNA-sequencing approaches, we detected large numbers of 3´ ends in 5´ UTRs and open reading frames (ORFs), suggesting extensive regulation by premature transcription termination. We documented regulation for multiple transcripts, including spermidine induction involving Rho and translation of an upstream ORF for an mRNA encoding a spermidine efflux pump. In addition to discovering novel sites of regulation, we detected short, stable RNA fragments derived from 5´ UTRs and sequences internal to ORFs. Characterization of three of these transcripts, including an RNA internal to an essential cell division gene, revealed that they have independent functions as sRNA sponges. Thus, these data uncover an abundance of cis- and trans-acting RNA regulators in bacterial 5´ UTRs and internal to ORFs.
Keywords: E. coli; RNA sponges; RNA-mediated regulation; RNA-seq; Rho-dependent termination; base pairing sRNAs; infectious disease; microbiology; uORF.
Plain language summary
In most organisms, specific segments of a cell’s genetic information are copied to form single-stranded molecules of various sizes and purposes. Each of these RNA molecules, as they are known, is constructed as a chain that starts at the 5´ end and terminates at the 3´ end. Certain RNAs carry the information present in a gene, which provides the instructions that a cell needs to build proteins. Some, however, are ‘non-coding’ and instead act to fine-tune the activity of other RNAs. These regulatory RNAs can be separate from the RNAs they control, or they can be embedded in the very sequences they regulate; new evidence also shows that certain regulatory RNAs can act in both ways. Many regulatory RNAs are yet to be catalogued, even in simple, well-studied species such as the bacterium Escherichia coli. Here, Adams et al. aimed to better characterize the regulatory RNAs present in E. coli by mapping out the 3´ ends of every RNA molecule in the bacterium. This revealed many new regulatory RNAs and offered insights into where these sequences are located. For instance, the results show that several of these RNAs were embedded within RNA produced from larger genes. Some were nested in coding RNAs, and were parts of a longer RNA sequence that is adjacent to the protein coding segment. Others, however, were present within the instructions that code for a protein. The work by Adams et al. reveals that regulatory RNAs can be located in unexpected places, and provides a method for identifying them. This can be applied to other types of bacteria, in particular in species with few known RNA regulators.
Conflict of interest statement
PA, GB, CE, KC, NS, MM, RD No competing interests declared, GS Senior editor, eLife, JW Reviewing editor, eLife
Figures














Similar articles
-
Dynamic interactions between the RNA chaperone Hfq, small regulatory RNAs, and mRNAs in live bacterial cells.Elife. 2021 Feb 22;10:e64207. doi: 10.7554/eLife.64207. Elife. 2021. PMID: 33616037 Free PMC article.
-
Regulation of Transcription Termination of Small RNAs and by Small RNAs: Molecular Mechanisms and Biological Functions.Front Cell Infect Microbiol. 2019 Jun 12;9:201. doi: 10.3389/fcimb.2019.00201. eCollection 2019. Front Cell Infect Microbiol. 2019. PMID: 31249814 Free PMC article. Review.
-
Hfq CLASH uncovers sRNA-target interaction networks linked to nutrient availability adaptation.Elife. 2020 May 1;9:e54655. doi: 10.7554/eLife.54655. Elife. 2020. PMID: 32356726 Free PMC article.
-
Bacterial Noncoding RNAs Excised from within Protein-Coding Transcripts.mBio. 2018 Sep 25;9(5):e01730-18. doi: 10.1128/mBio.01730-18. mBio. 2018. PMID: 30254125 Free PMC article.
-
New aspects of RNA-based regulation by Hfq and its partner sRNAs.Curr Opin Microbiol. 2018 Apr;42:53-61. doi: 10.1016/j.mib.2017.10.014. Epub 2017 Nov 7. Curr Opin Microbiol. 2018. PMID: 29125938 Free PMC article. Review.
Cited by
-
Ribosome profiling reveals the fine-tuned response of Escherichia coli to mild and severe acid stress.mSystems. 2023 Dec 21;8(6):e0103723. doi: 10.1128/msystems.01037-23. Epub 2023 Nov 1. mSystems. 2023. PMID: 37909716 Free PMC article.
-
Hidden in plain sight: challenges in proteomics detection of small ORF-encoded polypeptides.Microlife. 2022 May 14;3:uqac005. doi: 10.1093/femsml/uqac005. eCollection 2022. Microlife. 2022. PMID: 37223358 Free PMC article.
-
Genetic evidence for functional diversification of gram-negative intermembrane phospholipid transporters.PLoS Genet. 2024 Jun 24;20(6):e1011335. doi: 10.1371/journal.pgen.1011335. eCollection 2024 Jun. PLoS Genet. 2024. PMID: 38913742 Free PMC article.
-
TRS: a method for determining transcript termini from RNAtag-seq sequencing data.Nat Commun. 2023 Nov 29;14(1):7843. doi: 10.1038/s41467-023-43534-2. Nat Commun. 2023. PMID: 38030608 Free PMC article.
-
High-Resolution Small RNAs Landscape Provides Insights into Alkane Adaptation in the Marine Alkane-Degrader Alcanivorax dieselolei B-5.Int J Mol Sci. 2022 Dec 15;23(24):15995. doi: 10.3390/ijms232415995. Int J Mol Sci. 2022. PMID: 36555635 Free PMC article.
References
-
- Adams PP, Flores Avile C, Popitsch N, Bilusic I, Schroeder R, Lybecker M, Jewett MW. In vivo expression technology and 5' end mapping of the Borrelia burgdorferi transcriptome identify novel RNAs expressed during mammalian infection. Nucleic Acids Research. 2017;45:775–792. doi: 10.1093/nar/gkw1180. - DOI - PMC - PubMed
-
- Adams PP. termseq-peaks. swh:1:dir:1b9837a92dc90b4ea5d388fff43f5467a21c3b6eSoftware Heritage. 2020 https://archive.softwareheritage.org/swh:1:dir:1b9837a92dc90b4ea5d388fff...
-
- Apfel CM, Takács B, Fountoulakis M, Stieger M, Keck W. Use of genomics to identify bacterial undecaprenyl pyrophosphate synthetase: cloning, expression, and characterization of the essential uppS gene. Journal of Bacteriology. 1999;181:483–492. doi: 10.1128/JB.181.2.483-492.1999. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
