

Giant lungfish genome elucidates the conquest of land by vertebrates
- PMID: 33461212
- PMCID: PMC7875771
- DOI: 10.1038/s41586-021-03198-8
Giant lungfish genome elucidates the conquest of land by vertebrates
Abstract
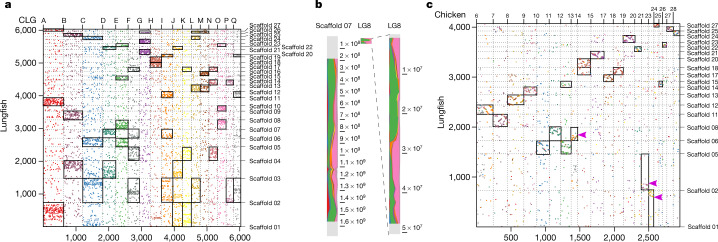
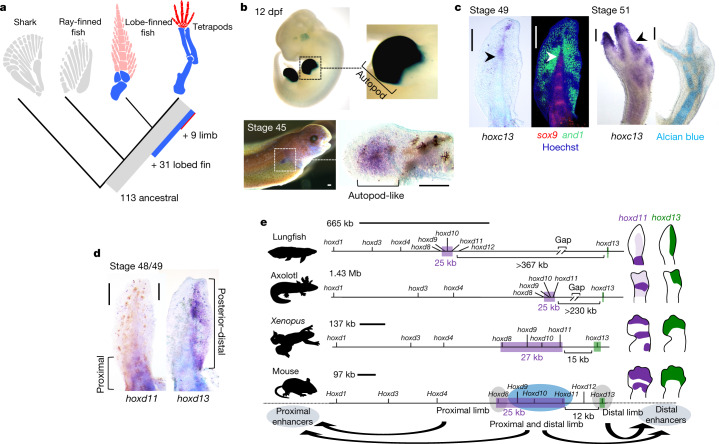
Lungfishes belong to lobe-fined fish (Sarcopterygii) that, in the Devonian period, 'conquered' the land and ultimately gave rise to all land vertebrates, including humans1-3. Here we determine the chromosome-quality genome of the Australian lungfish (Neoceratodus forsteri), which is known to have the largest genome of any animal. The vast size of this genome, which is about 14× larger than that of humans, is attributable mostly to huge intergenic regions and introns with high repeat content (around 90%), the components of which resemble those of tetrapods (comprising mainly long interspersed nuclear elements) more than they do those of ray-finned fish. The lungfish genome continues to expand independently (its transposable elements are still active), through mechanisms different to those of the enormous genomes of salamanders. The 17 fully assembled lungfish macrochromosomes maintain synteny to other vertebrate chromosomes, and all microchromosomes maintain conserved ancient homology with the ancestral vertebrate karyotype. Our phylogenomic analyses confirm previous reports that lungfish occupy a key evolutionary position as the closest living relatives to tetrapods4,5, underscoring the importance of lungfish for understanding innovations associated with terrestrialization. Lungfish preadaptations to living on land include the gain of limb-like expression in developmental genes such as hoxc13 and sall1 in their lobed fins. Increased rates of evolution and the duplication of genes associated with obligate air-breathing, such as lung surfactants and the expansion of odorant receptor gene families (which encode proteins involved in detecting airborne odours), contribute to the tetrapod-like biology of lungfishes. These findings advance our understanding of this major transition during vertebrate evolution.
Conflict of interest statement
The authors declare no competing interests.
Figures












Comment in
-
Giant genomes of lungfish.Nat Rev Genet. 2021 Apr;22(4):199. doi: 10.1038/s41576-021-00337-9. Nat Rev Genet. 2021. PMID: 33597743 No abstract available.
References
-
- Clack, J., Sharp, E. & Long, J. in The Biology of Lungfishes (eds Jorgensen, J. M. & Joss, J.) 1–42 (CRC, 2011).
-
- Kemp A. The biology of the Australian lungfish, Neoceratodus forsteri (Krefft 1870) J. Morphol. 1986;190:181–198.
-
- Carroll, R. L. Vertebrate Paleontology and Evolution (W. H. Freeman, 1988).
-
- Irisarri I, Meyer A. The identification of the closest living relative(s) of tetrapods: phylogenomic lessons for resolving short ancient internodes. Syst. Biol. 2016;65:1057–1075. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
