

A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy
- PMID: 33462228
- PMCID: PMC7814014
- DOI: 10.1038/s41467-020-20658-3
A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy
Abstract
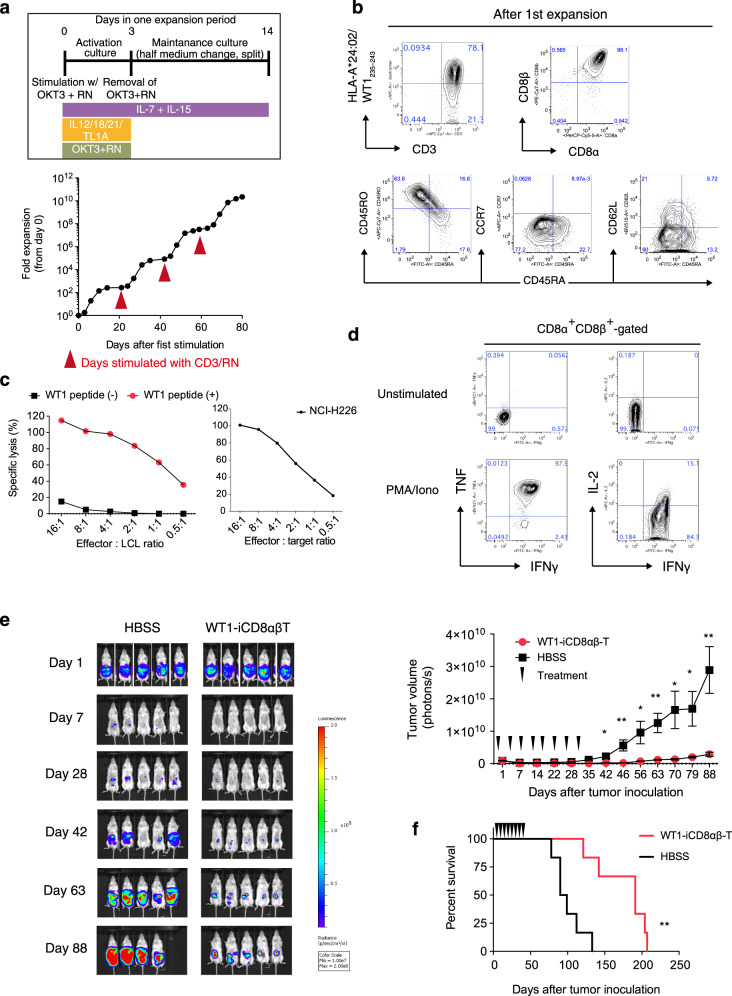
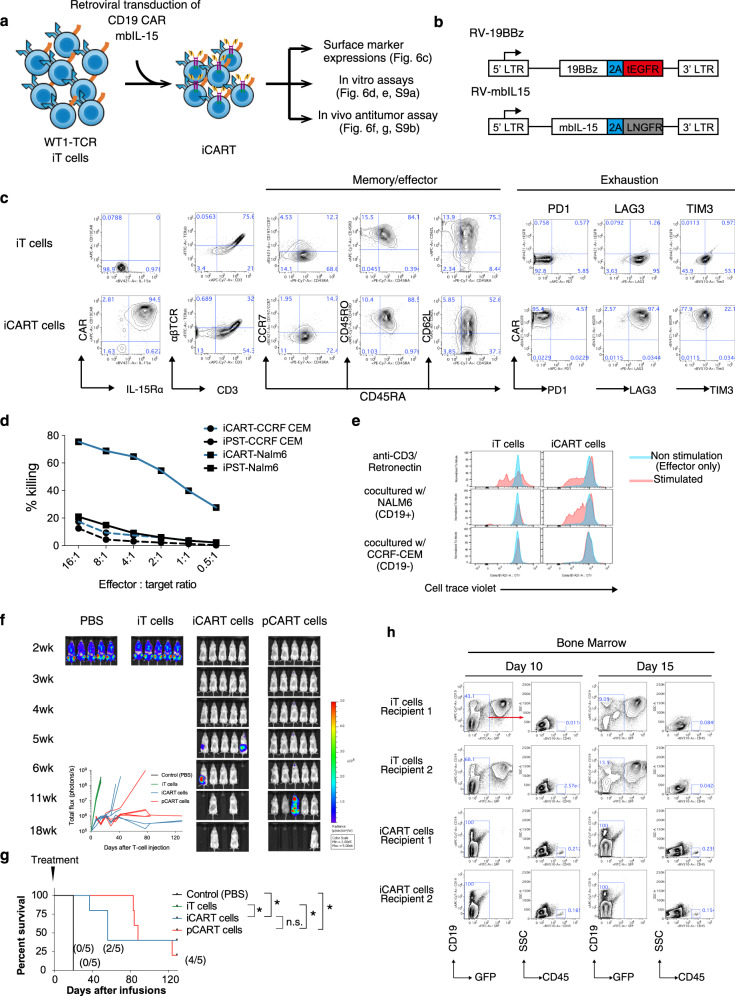
Clinical successes demonstrated by chimeric antigen receptor T-cell immunotherapy have facilitated further development of T-cell immunotherapy against wide variety of diseases. One approach is the development of "off-the-shelf" T-cell sources. Technologies to generate T-cells from pluripotent stem cells (PSCs) may offer platforms to produce "off-the-shelf" and synthetic allogeneic T-cells. However, low differentiation efficiency and poor scalability of current methods may compromise their utilities. Here we show improved differentiation efficiency of T-cells from induced PSCs (iPSCs) derived from an antigen-specific cytotoxic T-cell clone, or from T-cell receptor (TCR)-transduced iPSCs, as starting materials. We additionally describe feeder-free differentiation culture systems that span from iPSC maintenance to T-cell proliferation phases, enabling large-scale regenerated T-cell production. Moreover, simultaneous addition of SDF1α and a p38 inhibitor during T-cell differentiation enhances T-cell commitment. The regenerated T-cells show TCR-dependent functions in vitro and are capable of in vivo anti-tumor activity. This system provides a platform to generate a large number of regenerated T-cells for clinical application and investigate human T-cell differentiation and biology.
Conflict of interest statement
The authors declare the following financial competing interests. S.K. is a founder, shareholder, and chief scientific officer at Thyas Co., Ltd. and received research fundings from Takeda Pharmaceutical Co., Ltd., Kyowa Hakko Kirin Co., Ltd., Sumitomo Chemical Co., Ltd., and Thyas Co., Ltd. S.A., M.K., T. Sato., Y.B., T.S., K.N., M.T., Y. Kassai, and A.H. are employees of Takeda Pharmaceutical Co. Ltd. Y.Y. is an employee of Thyas Co. Ltd. The remaining authors declare no competing financial interests. These authors and all other authors declare no other competing interests.
Figures
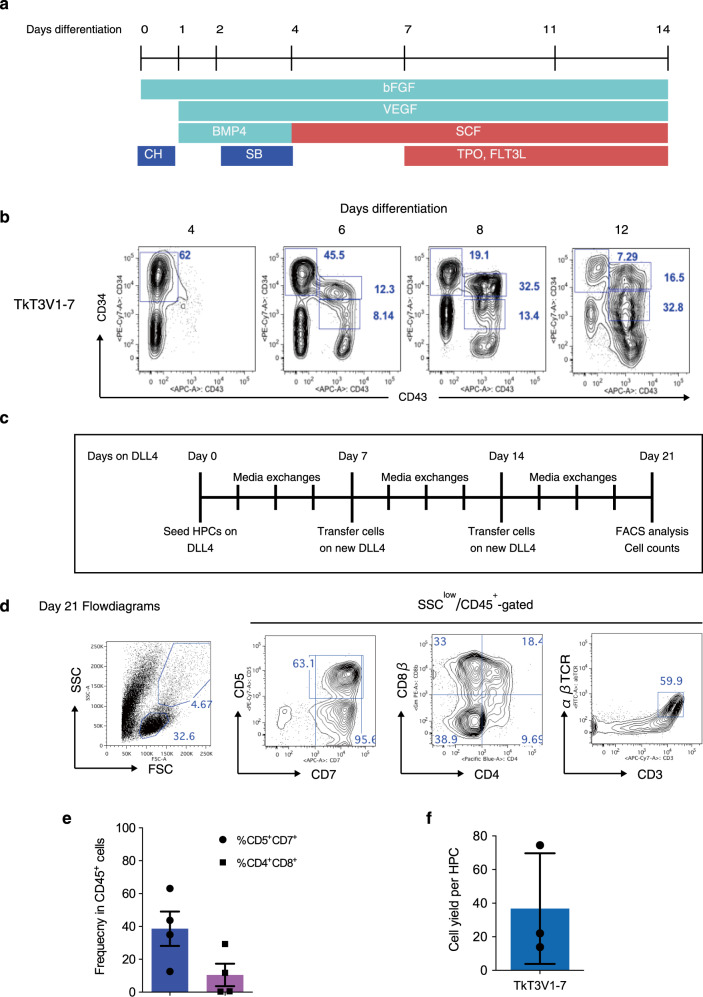
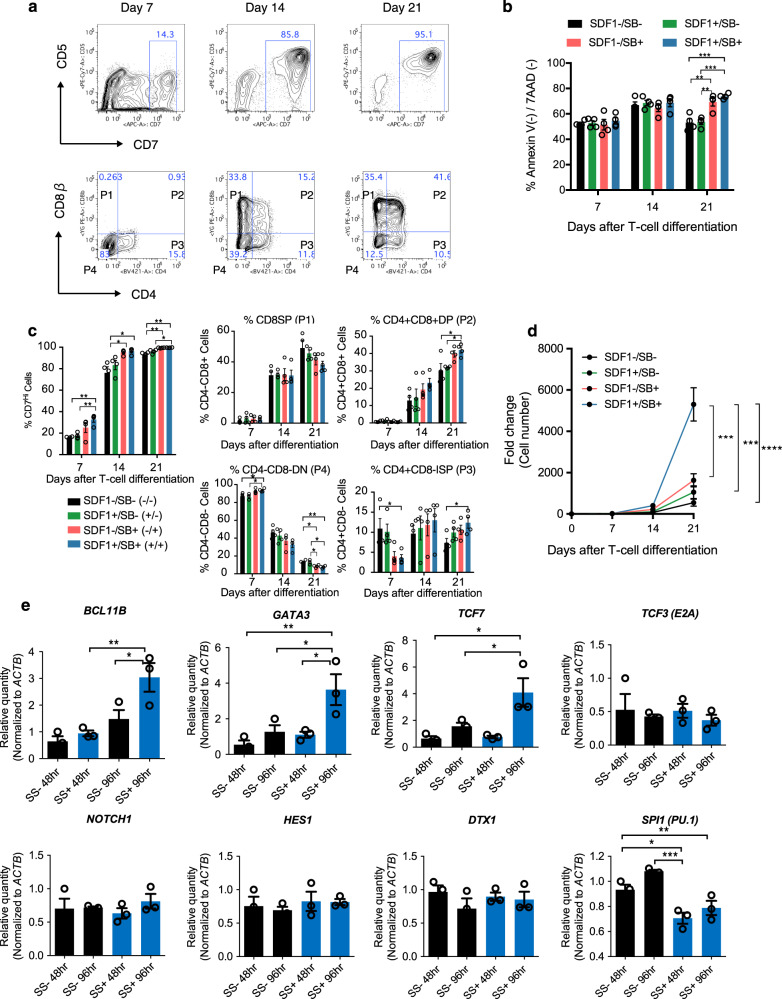
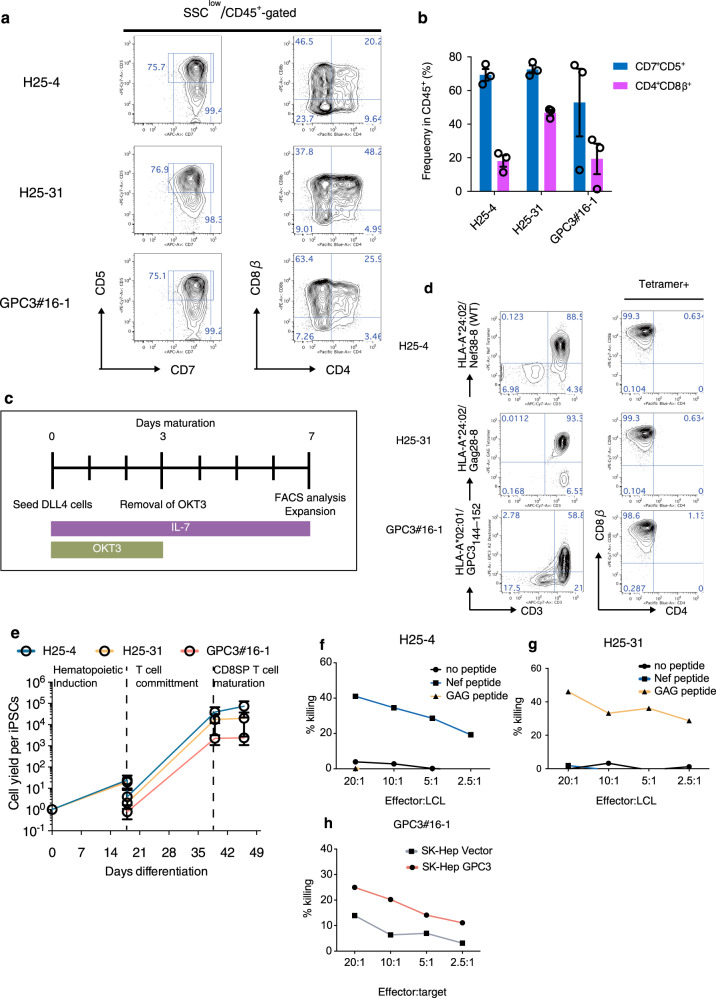
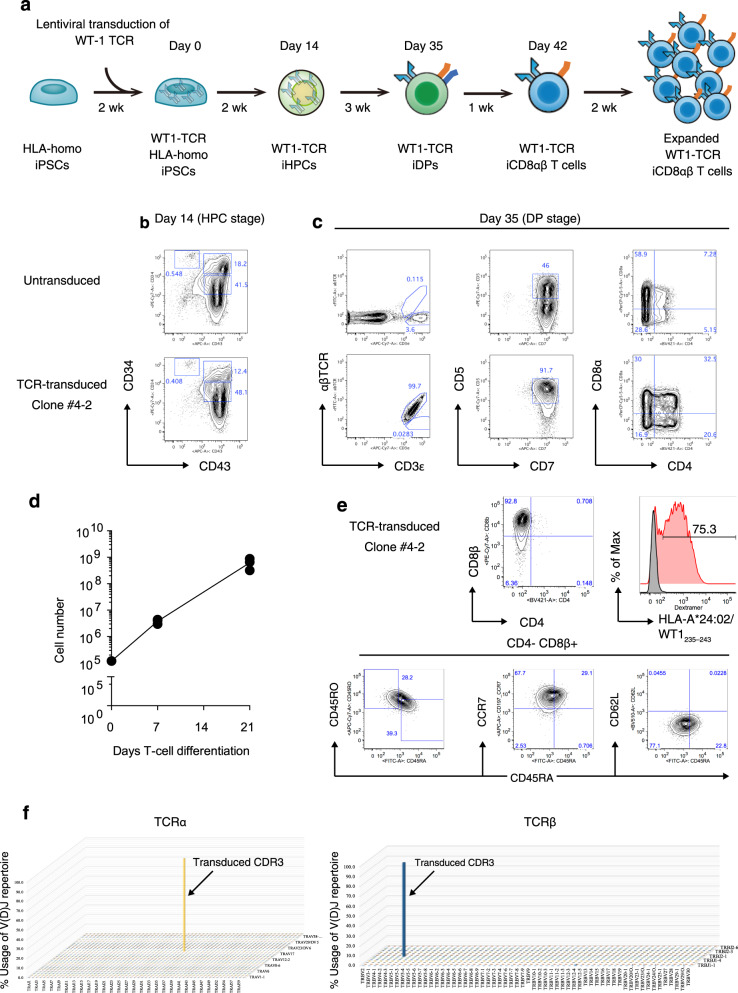
References
-
- Kennedy M, et al. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. CellReports. 2012;2:1722–1735. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
