

Core transcription regulatory circuitry orchestrates corneal epithelial homeostasis
- PMID: 33462242
- PMCID: PMC7814021
- DOI: 10.1038/s41467-020-20713-z
Core transcription regulatory circuitry orchestrates corneal epithelial homeostasis
Abstract
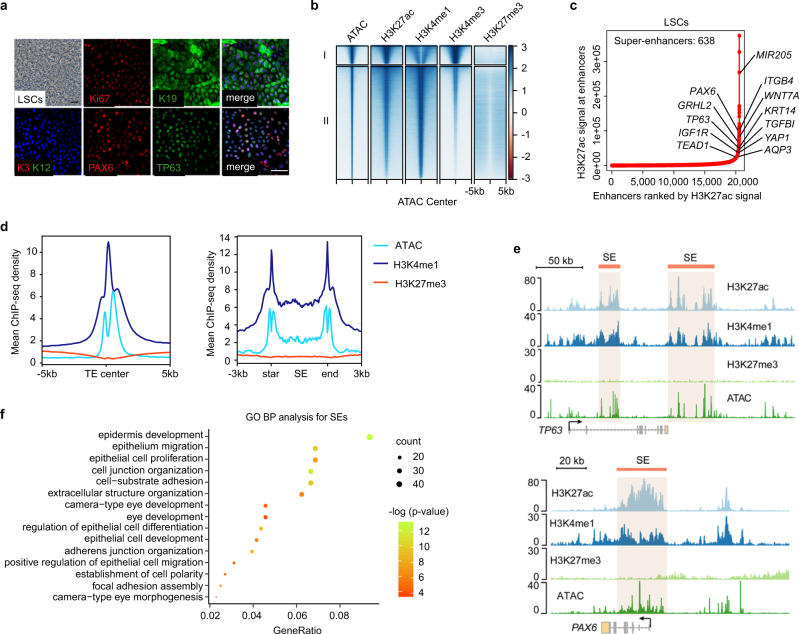
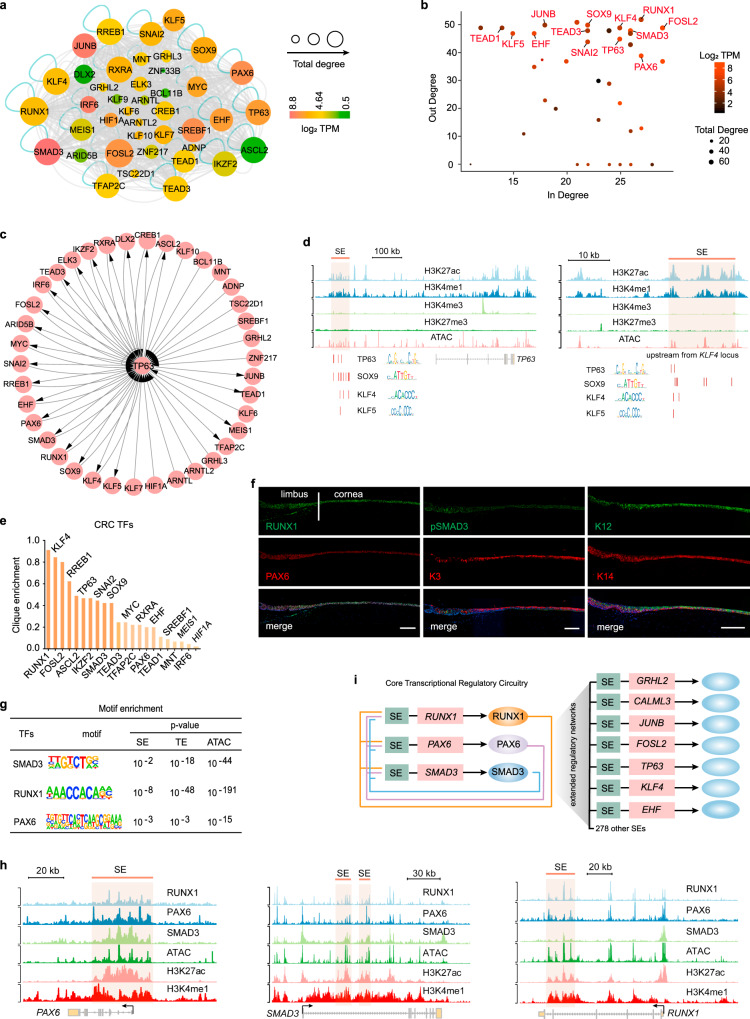
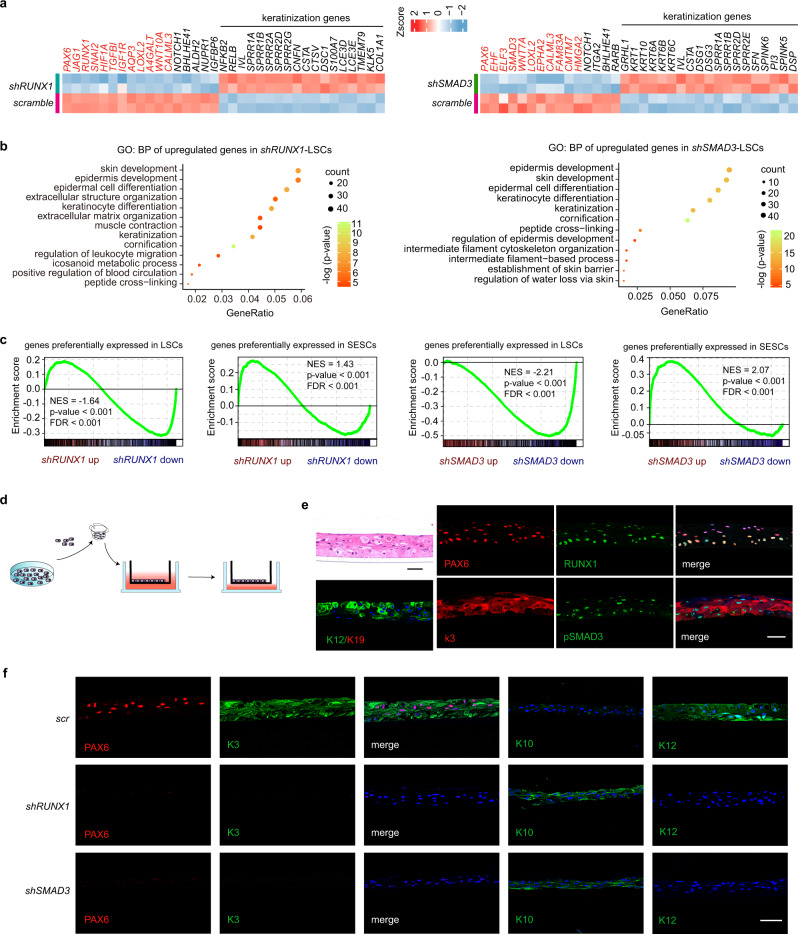
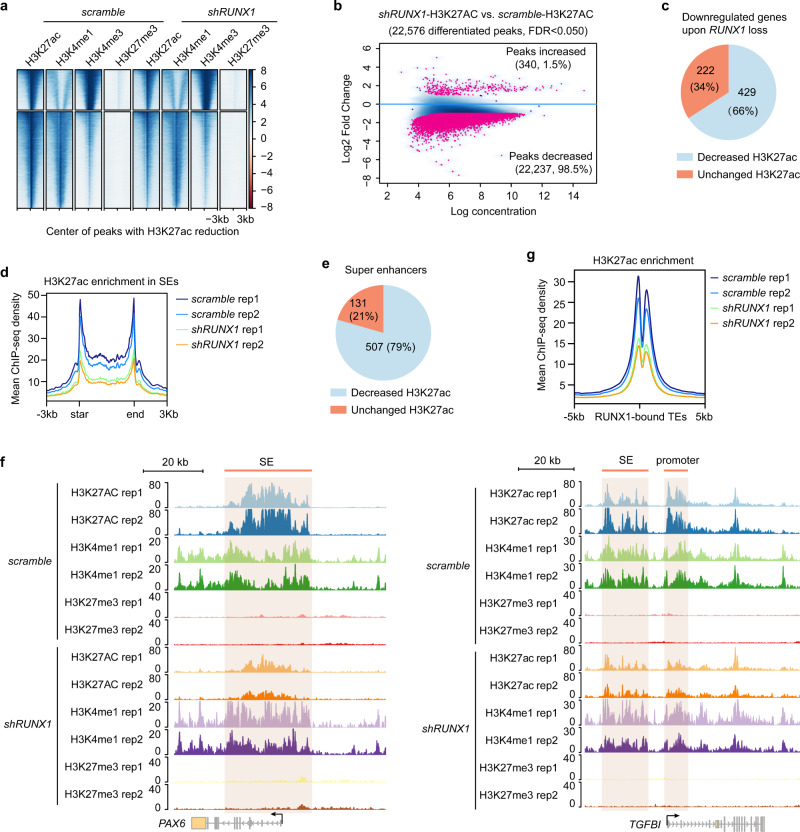
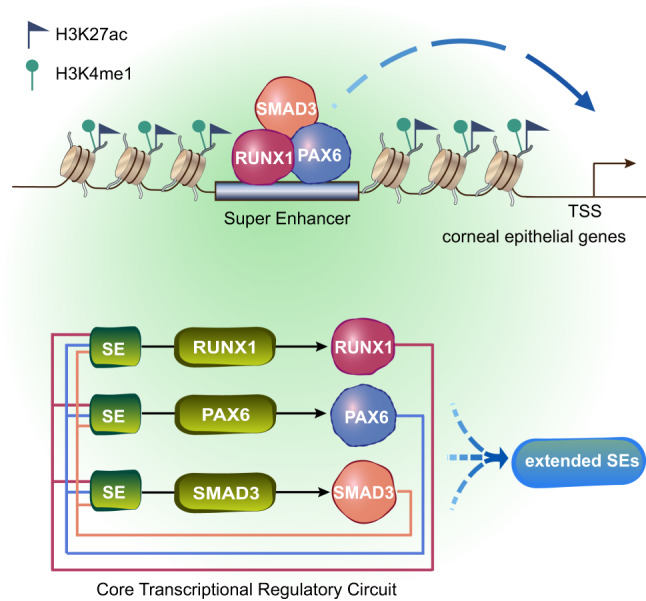
Adult stem cell identity, plasticity, and homeostasis are precisely orchestrated by lineage-restricted epigenetic and transcriptional regulatory networks. Here, by integrating super-enhancer and chromatin accessibility landscapes, we delineate core transcription regulatory circuitries (CRCs) of limbal stem/progenitor cells (LSCs) and find that RUNX1 and SMAD3 are required for maintenance of corneal epithelial identity and homeostasis. RUNX1 or SMAD3 depletion inhibits PAX6 and induces LSCs to differentiate into epidermal-like epithelial cells. RUNX1, PAX6, and SMAD3 (RPS) interact with each other and synergistically establish a CRC to govern the lineage-specific cis-regulatory atlas. Moreover, RUNX1 shapes LSC chromatin architecture via modulating H3K27ac deposition. Disturbance of RPS cooperation results in cell identity switching and dysfunction of the corneal epithelium, which is strongly linked to various human corneal diseases. Our work highlights CRC TF cooperativity for establishment of stem cell identity and lineage commitment, and provides comprehensive regulatory principles for human stratified epithelial homeostasis and pathogenesis.
Conflict of interest statement
The authors declare no competing interests.
Figures


Similar articles
-
PAX6-WNK2 Axis Governs Corneal Epithelial Homeostasis.Invest Ophthalmol Vis Sci. 2024 Oct 1;65(12):40. doi: 10.1167/iovs.65.12.40. Invest Ophthalmol Vis Sci. 2024. PMID: 39453672 Free PMC article.
-
WNT7A and PAX6 define corneal epithelium homeostasis and pathogenesis.Nature. 2014 Jul 17;511(7509):358-61. doi: 10.1038/nature13465. Epub 2014 Jul 2. Nature. 2014. PMID: 25030175 Free PMC article.
-
Human corneal epithelial subpopulations: oxygen dependent ex vivo expansion and transcriptional profiling.Acta Ophthalmol. 2013 Jun;91 Thesis 4:1-34. doi: 10.1111/aos.12157. Acta Ophthalmol. 2013. PMID: 23732018
-
Corneal epithelial stem cells: characterization, culture and transplantation.Regen Med. 2006 Jan;1(1):29-44. doi: 10.2217/17460751.1.1.29. Regen Med. 2006. PMID: 17465818 Review.
-
Niche regulation of corneal epithelial stem cells at the limbus.Cell Res. 2007 Jan;17(1):26-36. doi: 10.1038/sj.cr.7310137. Cell Res. 2007. PMID: 17211449 Free PMC article. Review.
Cited by
-
PAX6-WNK2 Axis Governs Corneal Epithelial Homeostasis.Invest Ophthalmol Vis Sci. 2024 Oct 1;65(12):40. doi: 10.1167/iovs.65.12.40. Invest Ophthalmol Vis Sci. 2024. PMID: 39453672 Free PMC article.
-
RUNX1 colludes with NOTCH1 to reprogram chromatin in T cell acute lymphoblastic leukemia.iScience. 2023 May 3;26(6):106795. doi: 10.1016/j.isci.2023.106795. eCollection 2023 Jun 16. iScience. 2023. PMID: 37213235 Free PMC article.
-
Time-course swRNA-seq uncovers a hierarchical gene regulatory network in controlling the response-repair-remodeling after wounding.Commun Biol. 2024 Jun 6;7(1):694. doi: 10.1038/s42003-024-06352-w. Commun Biol. 2024. PMID: 38844830 Free PMC article.
-
Prediction of cell states and key transcription factors of the human cornea through integrated single-cell omics analyses.PNAS Nexus. 2025 Jul 29;4(8):pgaf235. doi: 10.1093/pnasnexus/pgaf235. eCollection 2025 Aug. PNAS Nexus. 2025. PMID: 40838019 Free PMC article.
-
Toward Corneal Limbus In Vitro Model: Regulation of hPSC-LSC Phenotype by Matrix Stiffness and Topography During Cell Differentiation Process.Adv Healthc Mater. 2023 Nov;12(29):e2301396. doi: 10.1002/adhm.202301396. Epub 2023 Jul 21. Adv Healthc Mater. 2023. PMID: 37449943 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
