

Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes
- PMID: 33462507
- PMCID: PMC8122019
- DOI: 10.1038/s41587-020-00795-2
Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes
Abstract
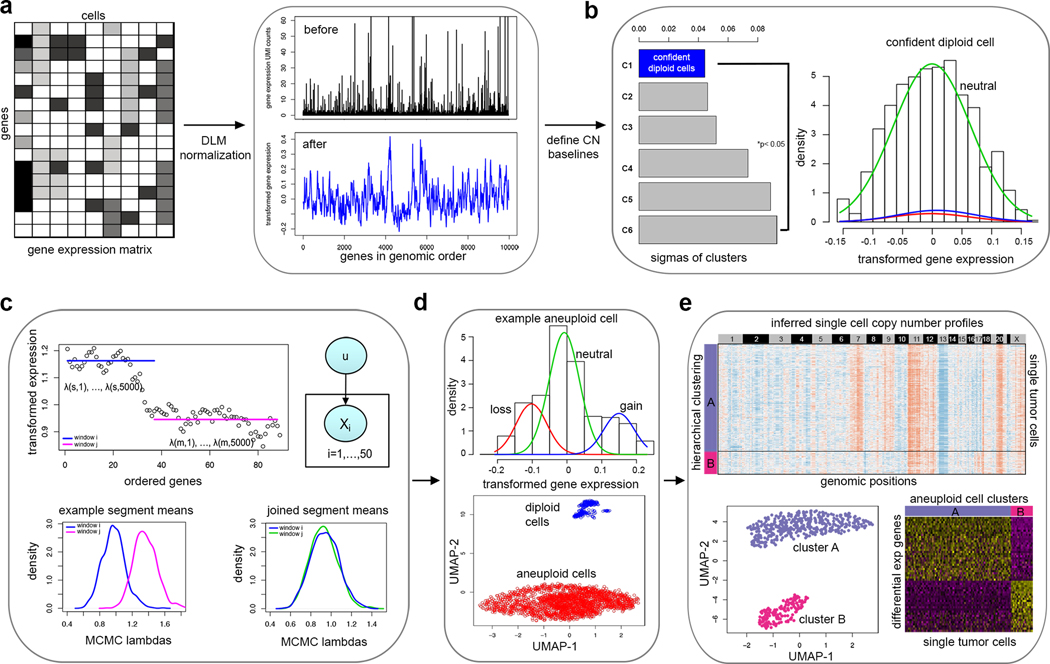
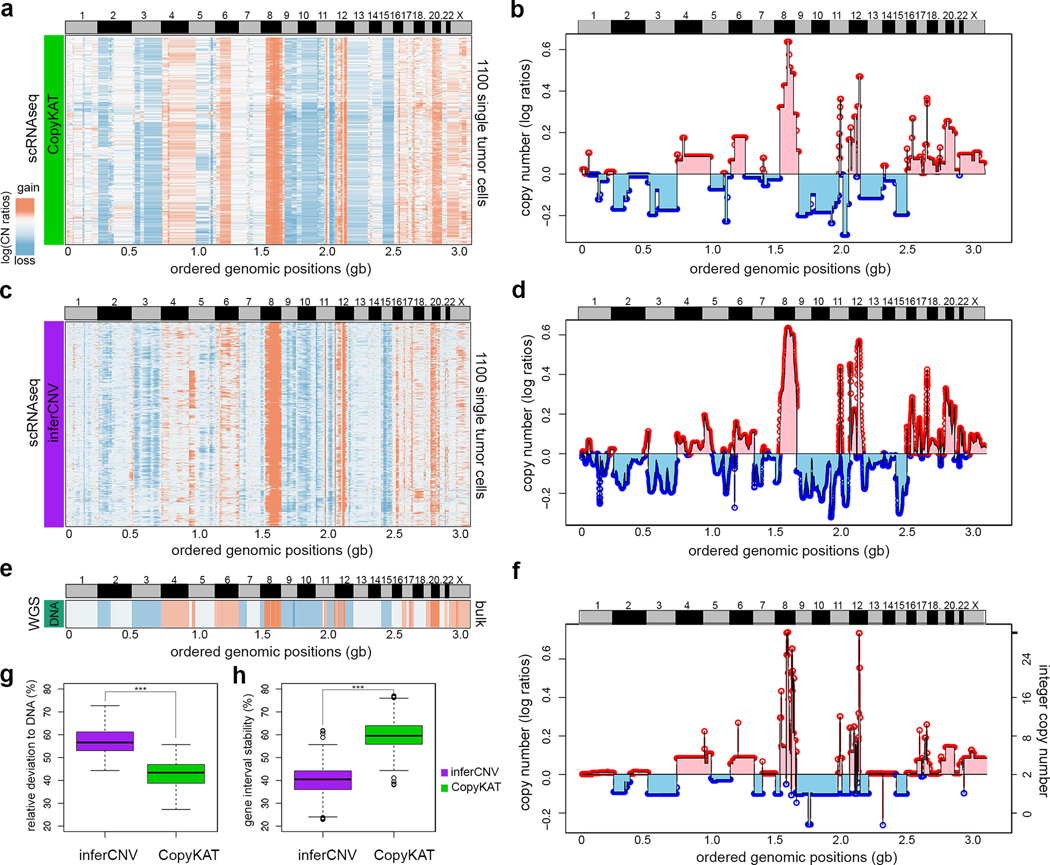
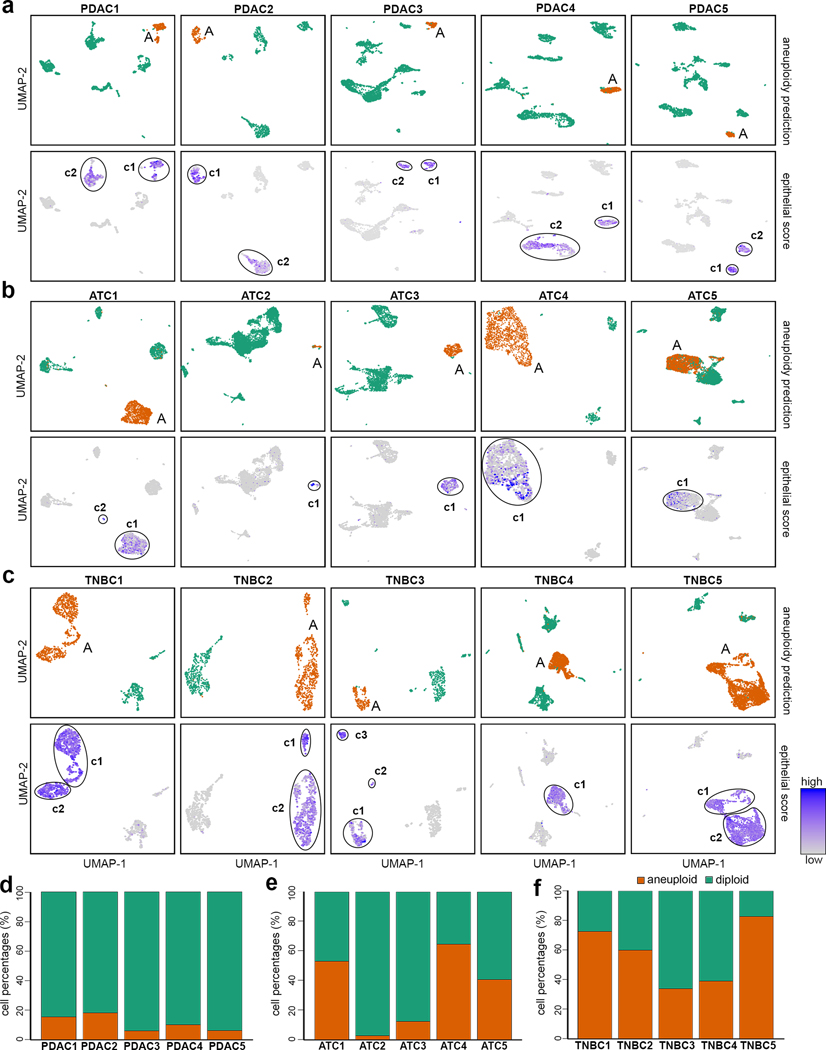
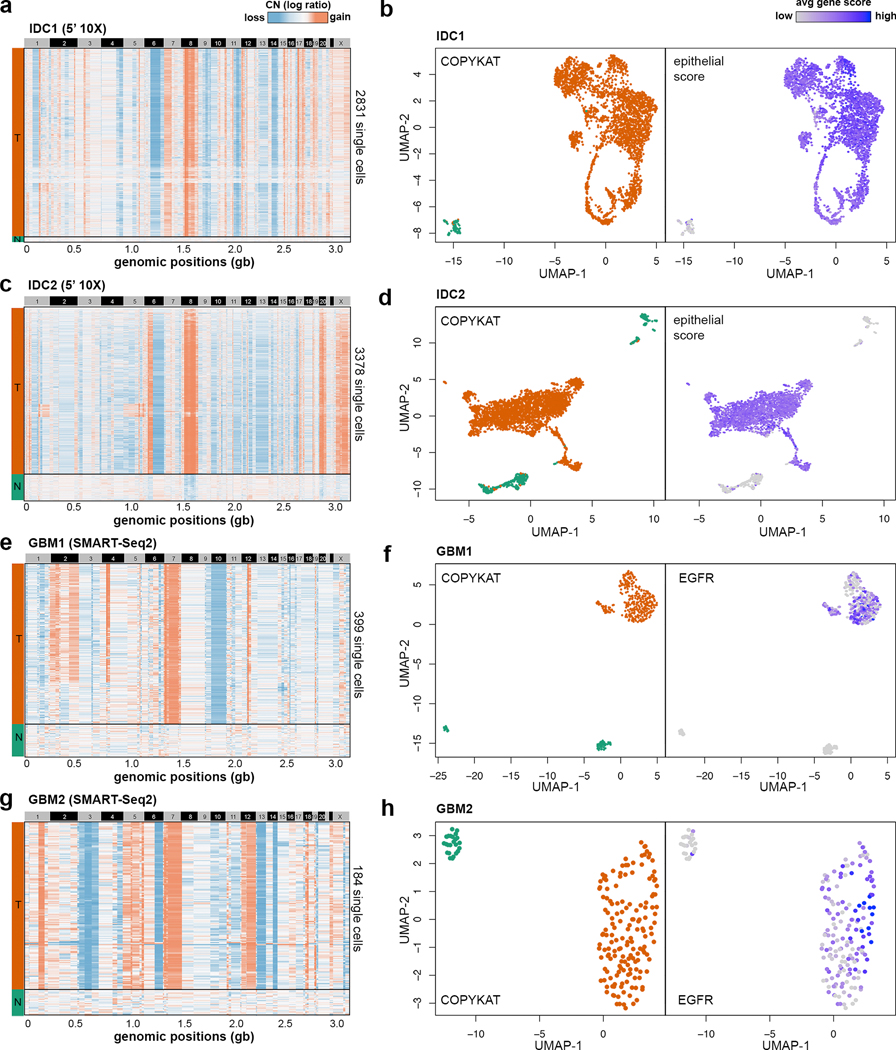
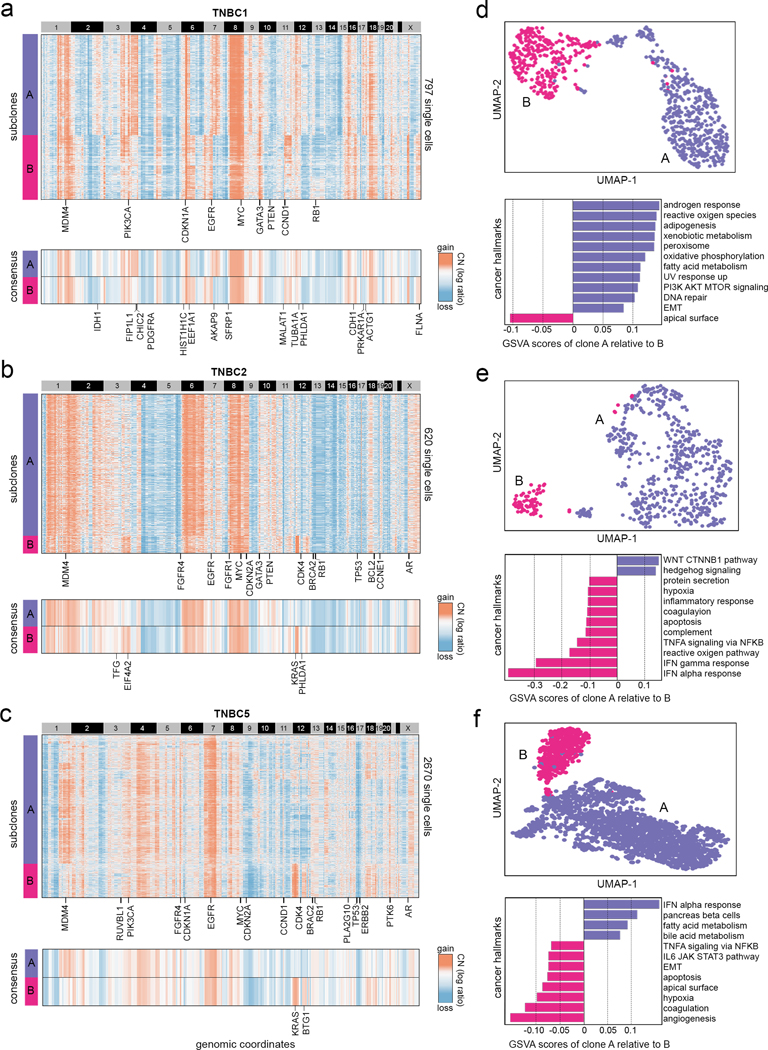
Single-cell transcriptomic analysis is widely used to study human tumors. However, it remains challenging to distinguish normal cell types in the tumor microenvironment from malignant cells and to resolve clonal substructure within the tumor. To address these challenges, we developed an integrative Bayesian segmentation approach called copy number karyotyping of aneuploid tumors (CopyKAT) to estimate genomic copy number profiles at an average genomic resolution of 5 Mb from read depth in high-throughput single-cell RNA sequencing (scRNA-seq) data. We applied CopyKAT to analyze 46,501 single cells from 21 tumors, including triple-negative breast cancer, pancreatic ductal adenocarcinoma, anaplastic thyroid cancer, invasive ductal carcinoma and glioblastoma, to accurately (98%) distinguish cancer cells from normal cell types. In three breast tumors, CopyKAT resolved clonal subpopulations that differed in the expression of cancer genes, such as KRAS, and signatures, including epithelial-to-mesenchymal transition, DNA repair, apoptosis and hypoxia. These data show that CopyKAT can aid in the analysis of scRNA-seq data in a variety of solid human tumors.
Figures
References
References (for main text only)
References (for Methods only)
-
- Martin AD, Quinn KM & Park JH MCMCpack: Markov Chain Monte Carlo in R.” Journal of Statistical Software. J Stat Softw 42, 22 (2011).
-
- Olshen AB, Venkatraman ES, Lucito R. & Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572 (2004). - PubMed
-
- Willenbrock H. & Fridlyand J. A comparison study: applying segmentation to array CGH data for downstream analyses. Bioinformatics 21, 4084–4091 (2005). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
