

Hepatocyte ATF3 protects against atherosclerosis by regulating HDL and bile acid metabolism
- PMID: 33462514
- PMCID: PMC7856821
- DOI: 10.1038/s42255-020-00331-1
Hepatocyte ATF3 protects against atherosclerosis by regulating HDL and bile acid metabolism
Abstract
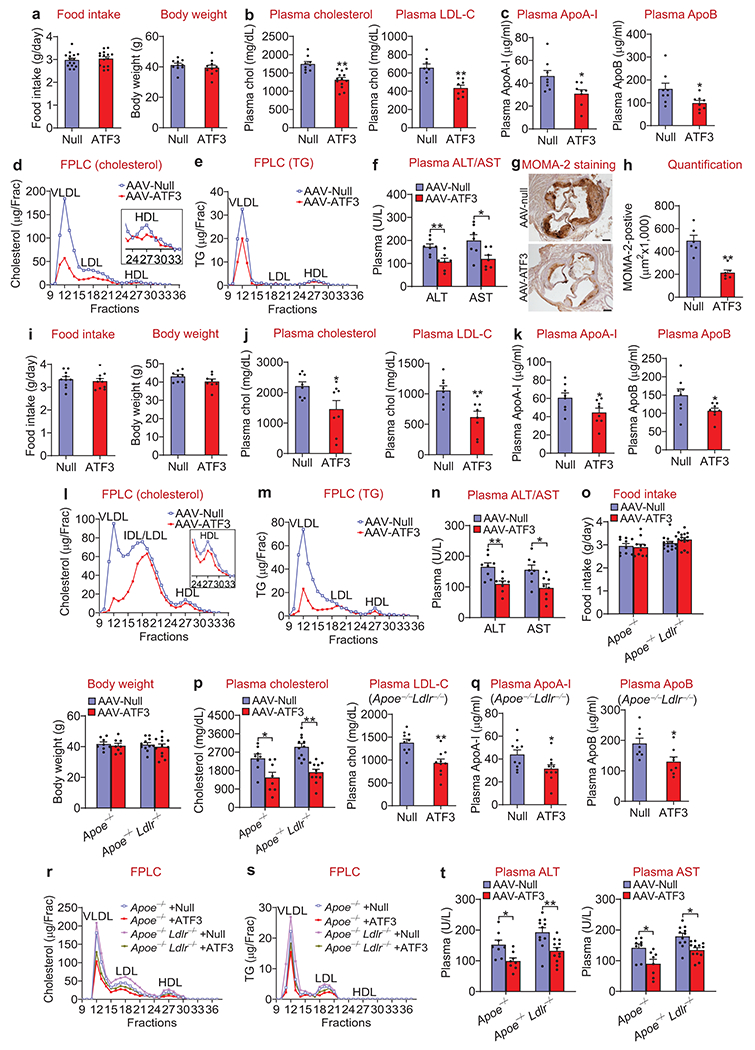
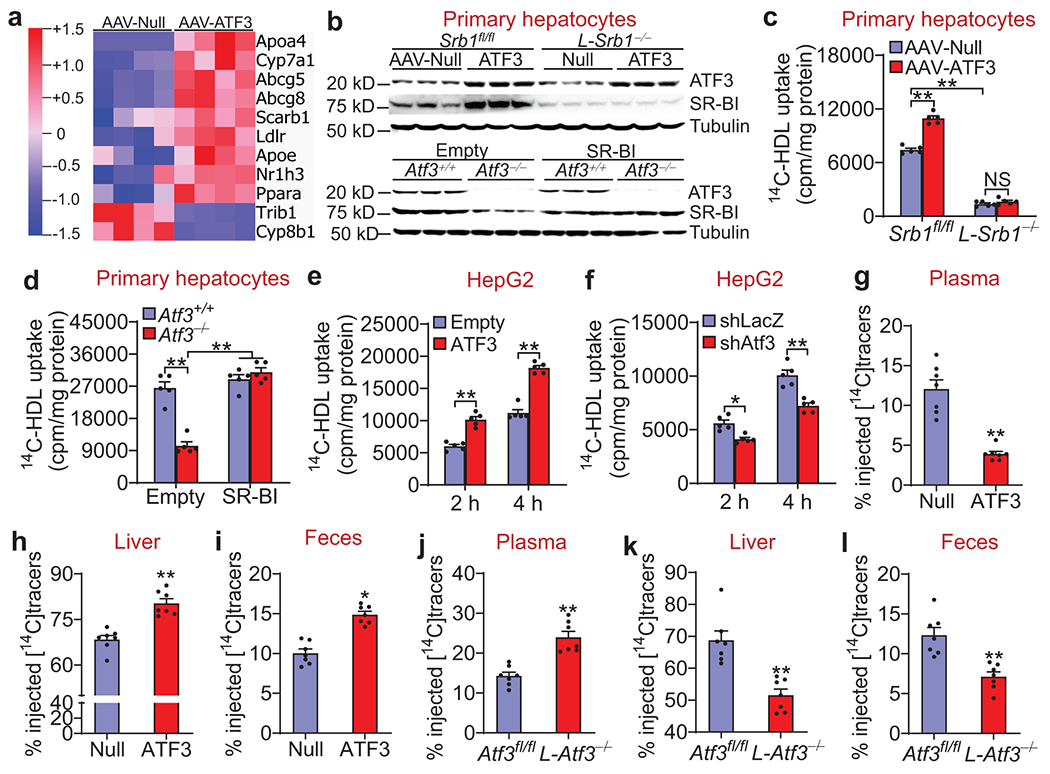
Activating transcription factor (ATF)3 is known to have an anti-inflammatory function, yet the role of hepatic ATF3 in lipoprotein metabolism or atherosclerosis remains unknown. Here we show that overexpression of human ATF3 in hepatocytes reduces the development of atherosclerosis in Western-diet-fed Ldlr-/- or Apoe-/- mice, whereas hepatocyte-specific ablation of Atf3 has the opposite effect. We further show that hepatic ATF3 expression is inhibited by hydrocortisone. Mechanistically, hepatocyte ATF3 enhances high-density lipoprotein (HDL) uptake, inhibits intestinal fat and cholesterol absorption and promotes macrophage reverse cholesterol transport by inducing scavenger receptor group B type 1 (SR-BI) and repressing cholesterol 12α-hydroxylase (CYP8B1) in the liver through its interaction with p53 and hepatocyte nuclear factor 4α, respectively. Our data demonstrate that hepatocyte ATF3 is a key regulator of HDL and bile acid metabolism and atherosclerosis.
Conflict of interest statement
Competing Interests
The authors declare no competing financial/non-financial interests.
Figures















References
-
- Braun A et al. Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ Res 90, 270–276 (2002). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
