

Comprehensive analysis of genomic diversity of SARS-CoV-2 in different geographic regions of India: an endeavour to classify Indian SARS-CoV-2 strains on the basis of co-existing mutations
- PMID: 33464421
- PMCID: PMC7814186
- DOI: 10.1007/s00705-020-04911-0
Comprehensive analysis of genomic diversity of SARS-CoV-2 in different geographic regions of India: an endeavour to classify Indian SARS-CoV-2 strains on the basis of co-existing mutations
Abstract
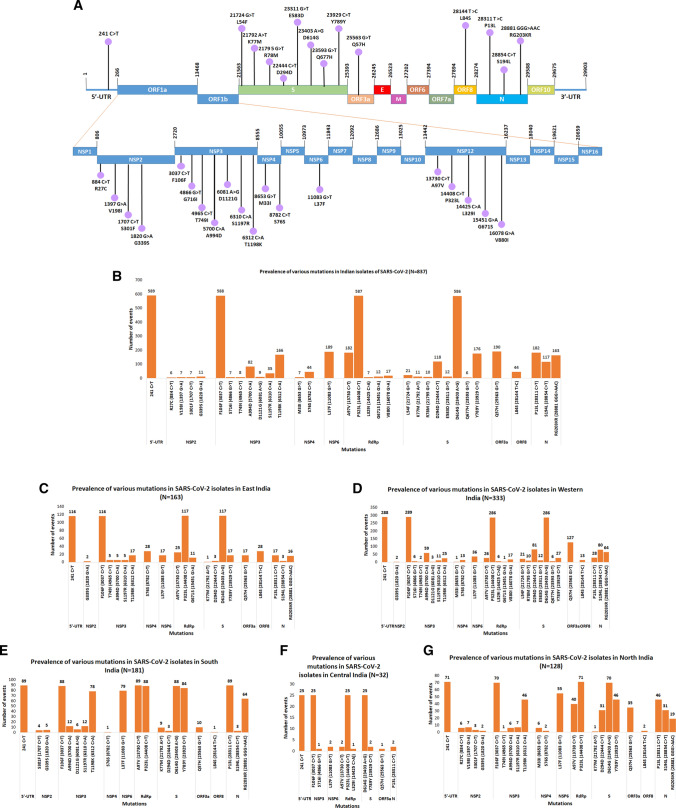
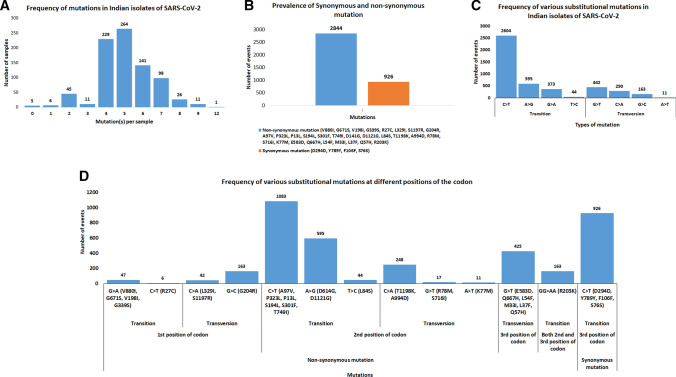
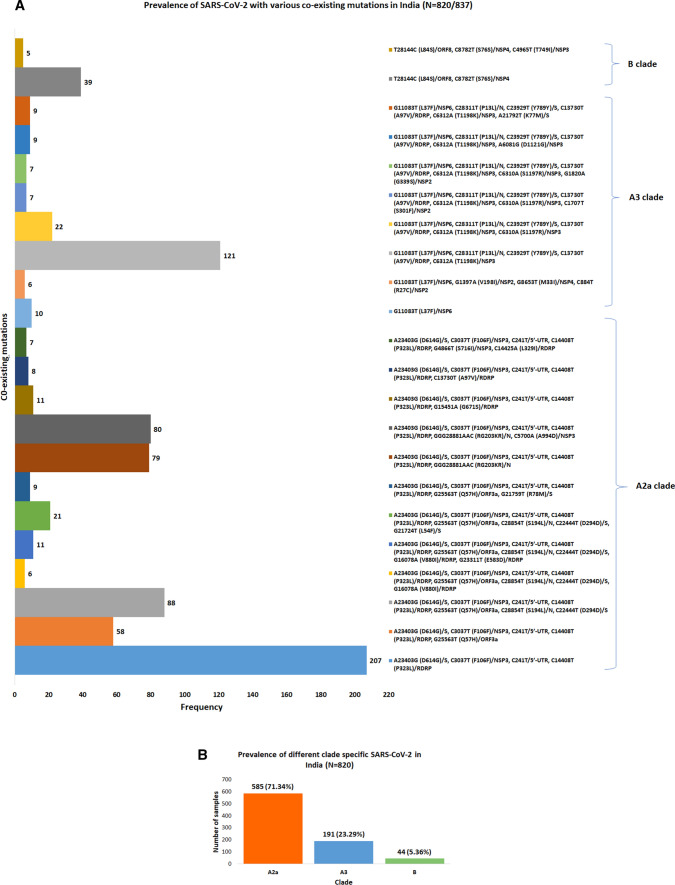
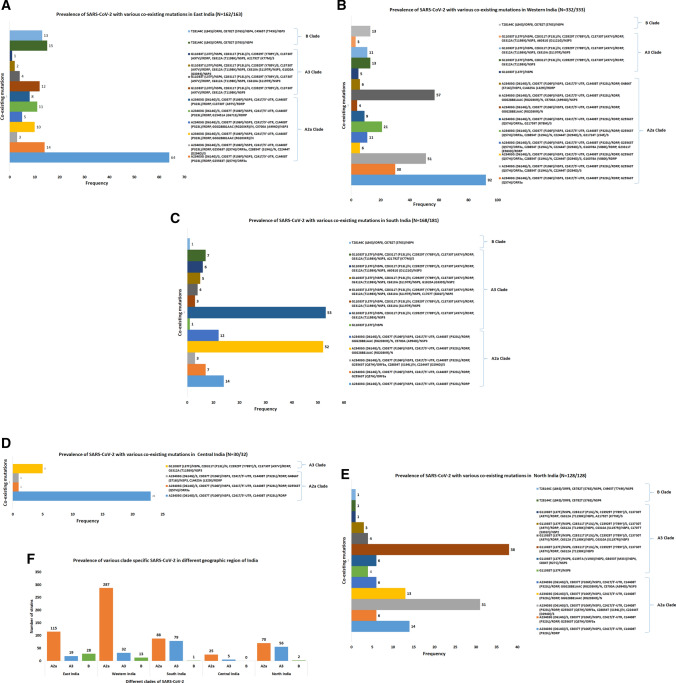
Accumulation of mutations within the genome is the primary driving force in viral evolution within an endemic setting. This inherent feature often leads to altered virulence, infectivity and transmissibility, and antigenic shifts to escape host immunity, which might compromise the efficacy of vaccines and antiviral drugs. Therefore, we carried out a genome-wide analysis of circulating SARS-CoV-2 strains to detect the emergence of novel co-existing mutations and trace their geographical distribution within India. Comprehensive analysis of whole genome sequences of 837 Indian SARS-CoV-2 strains revealed the occurrence of 33 different mutations, 18 of which were unique to India. Novel mutations were observed in the S glycoprotein (6/33), NSP3 (5/33), RdRp/NSP12 (4/33), NSP2 (2/33), and N (1/33). Non-synonymous mutations were found to be 3.07 times more prevalent than synonymous mutations. We classified the Indian isolates into 22 groups based on their co-existing mutations. Phylogenetic analysis revealed that the representative strains of each group were divided into various sub-clades within their respective clades, based on the presence of unique co-existing mutations. The A2a clade was found to be dominant in India (71.34%), followed by A3 (23.29%) and B (5.36%), but a heterogeneous distribution was observed among various geographical regions. The A2a clade was highly predominant in East India, Western India, and Central India, whereas the A2a and A3 clades were nearly equal in prevalence in South and North India. This study highlights the divergent evolution of SARS-CoV-2 strains and co-circulation of multiple clades in India. Monitoring of the emerging mutations will pave the way for vaccine formulation and the design of antiviral drugs.
Conflict of interest statement
The authors declare that no conflict of interest exists.
Figures

References
-
- Barr JN, Fearns R (2016) Genetic instability of RNA viruses. In: Genome stability. Academic Press, pp 21–35
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
