

Estimating FST and kinship for arbitrary population structures
- PMID: 33465078
- PMCID: PMC7846127
- DOI: 10.1371/journal.pgen.1009241
Estimating FST and kinship for arbitrary population structures
Abstract
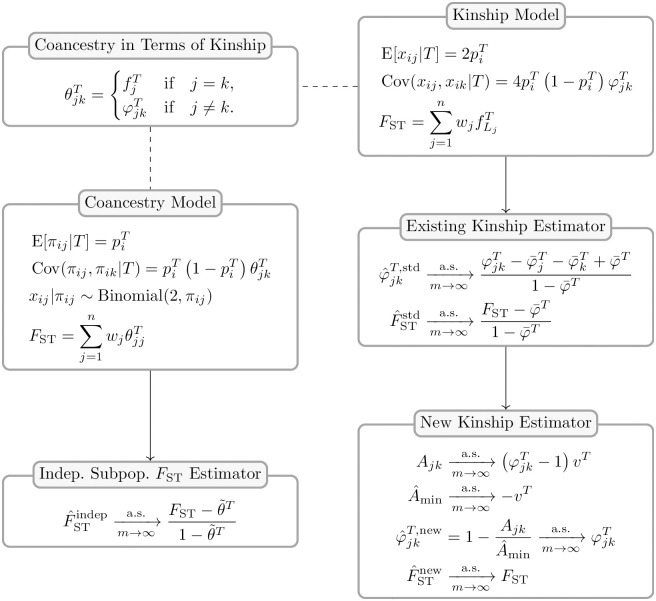
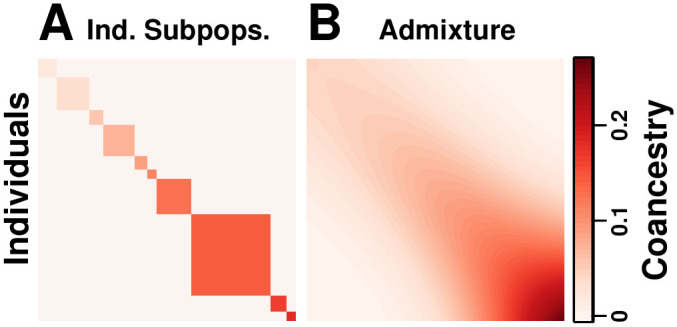
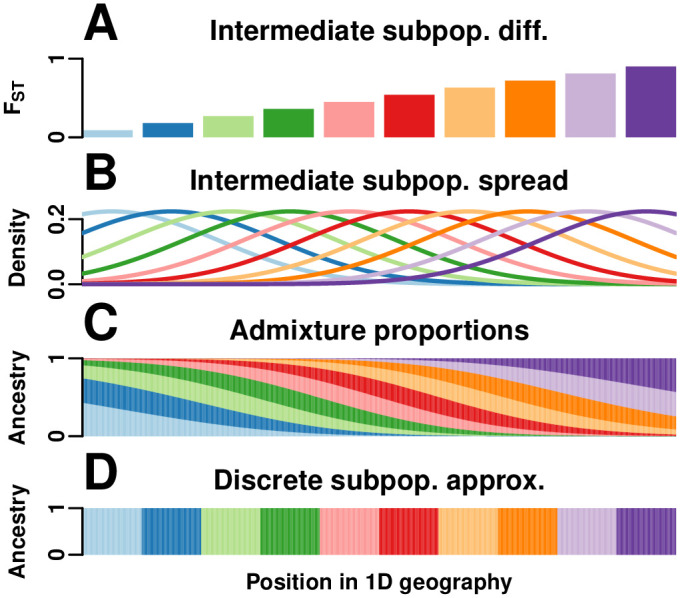
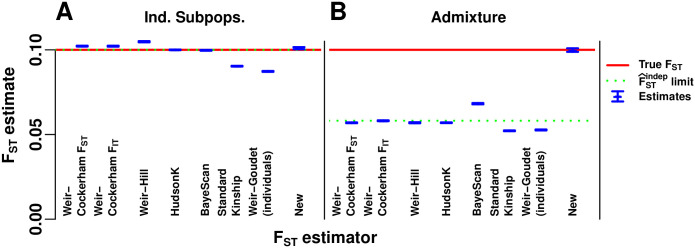
FST and kinship are key parameters often estimated in modern population genetics studies in order to quantitatively characterize structure and relatedness. Kinship matrices have also become a fundamental quantity used in genome-wide association studies and heritability estimation. The most frequently-used estimators of FST and kinship are method-of-moments estimators whose accuracies depend strongly on the existence of simple underlying forms of structure, such as the independent subpopulations model of non-overlapping, independently evolving subpopulations. However, modern data sets have revealed that these simple models of structure likely do not hold in many populations, including humans. In this work, we analyze the behavior of these estimators in the presence of arbitrarily-complex population structures, which results in an improved estimation framework specifically designed for arbitrary population structures. After generalizing the definition of FST to arbitrary population structures and establishing a framework for assessing bias and consistency of genome-wide estimators, we calculate the accuracy of existing FST and kinship estimators under arbitrary population structures, characterizing biases and estimation challenges unobserved under their originally-assumed models of structure. We then present our new approach, which consistently estimates kinship and FST when the minimum kinship value in the dataset is estimated consistently. We illustrate our results using simulated genotypes from an admixture model, constructing a one-dimensional geographic scenario that departs nontrivially from the independent subpopulations model. Our simulations reveal the potential for severe biases in estimates of existing approaches that are overcome by our new framework. This work may significantly improve future analyses that rely on accurate kinship and FST estimates.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
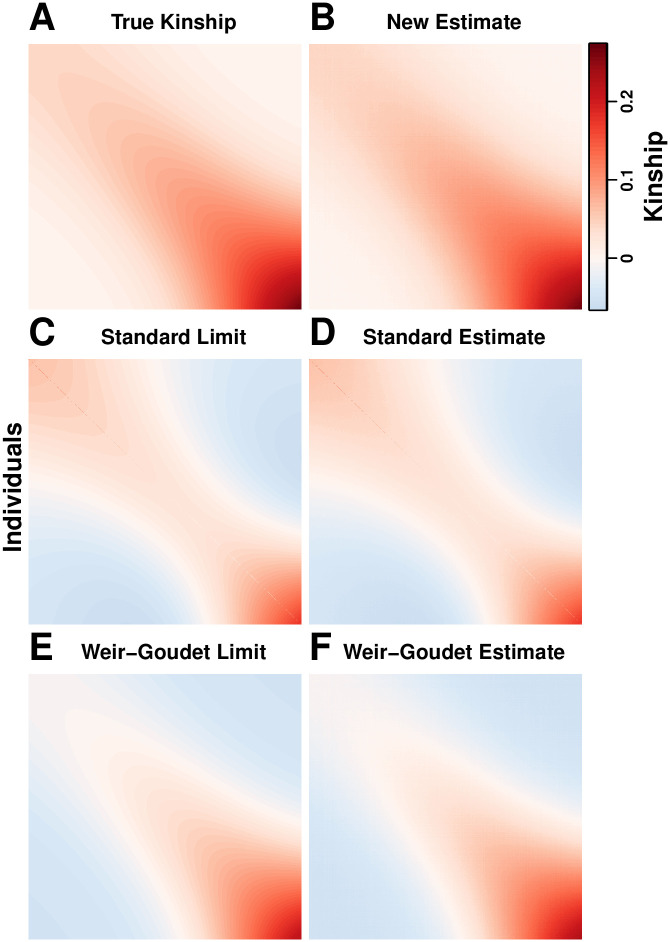
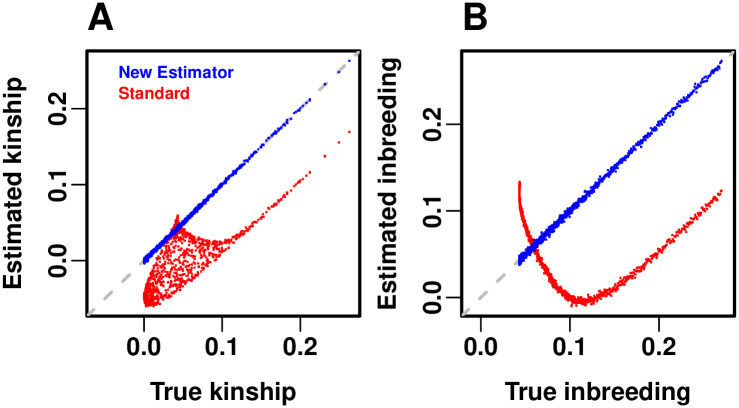
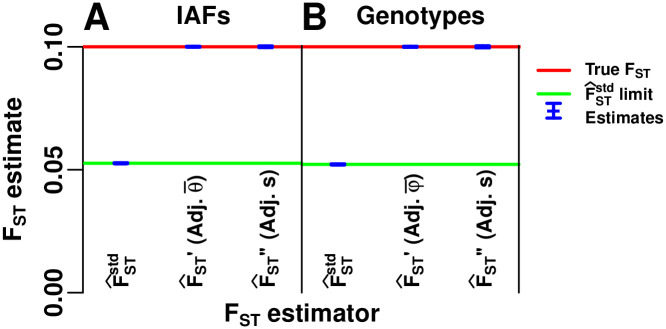
References
-
- Malécot G. Mathématiques de l’hérédité. Masson et Cie; 1948.
-
- Wright S. The genetical structure of populations. Ann Eugen. 1951;15(4):323–354. - PubMed
-
- Nicholson G, Smith AV, Jónsson F, Gústafsson O, Stefánsson K, Donnelly P. Assessing population differentiation and isolation from single-nucleotide polymorphism data. Journal of the Royal Statistical Society: Series B (Statistical Methodology). 2002;64(4):695–715. 10.1111/1467-9868.00357 - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
