

The coronavirus proofreading exoribonuclease mediates extensive viral recombination
- PMID: 33465137
- PMCID: PMC7846108
- DOI: 10.1371/journal.ppat.1009226
The coronavirus proofreading exoribonuclease mediates extensive viral recombination
Abstract
Recombination is proposed to be critical for coronavirus (CoV) diversity and emergence of SARS-CoV-2 and other zoonotic CoVs. While RNA recombination is required during normal CoV replication, the mechanisms and determinants of CoV recombination are not known. CoVs encode an RNA proofreading exoribonuclease (nsp14-ExoN) that is distinct from the CoV polymerase and is responsible for high-fidelity RNA synthesis, resistance to nucleoside analogues, immune evasion, and virulence. Here, we demonstrate that CoVs, including SARS-CoV-2, MERS-CoV, and the model CoV murine hepatitis virus (MHV), generate extensive and diverse recombination products during replication in culture. We show that the MHV nsp14-ExoN is required for native recombination, and that inactivation of ExoN results in decreased recombination frequency and altered recombination products. These results add yet another critical function to nsp14-ExoN, highlight the uniqueness of the evolved coronavirus replicase, and further emphasize nsp14-ExoN as a central, completely conserved, and vulnerable target for inhibitors and attenuation of SARS-CoV-2 and future emerging zoonotic CoVs.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
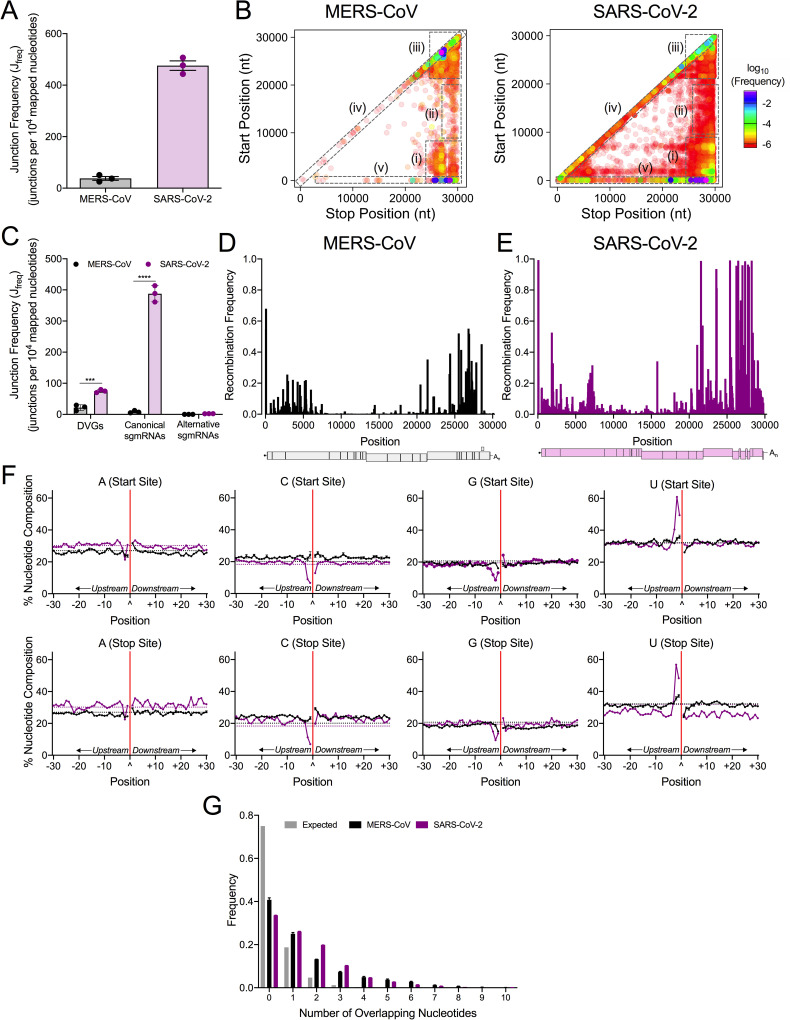
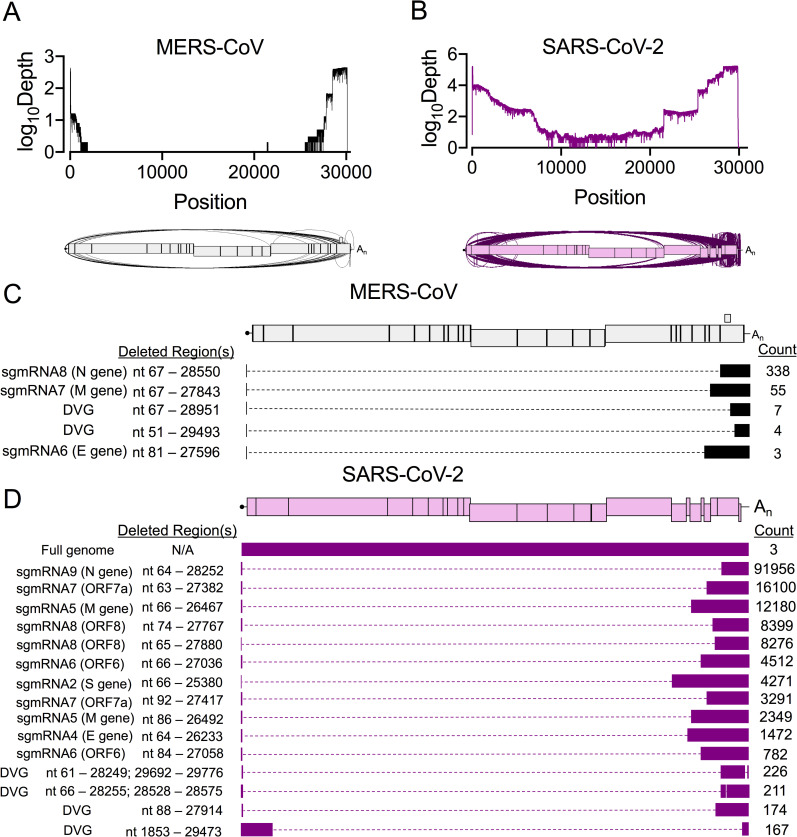
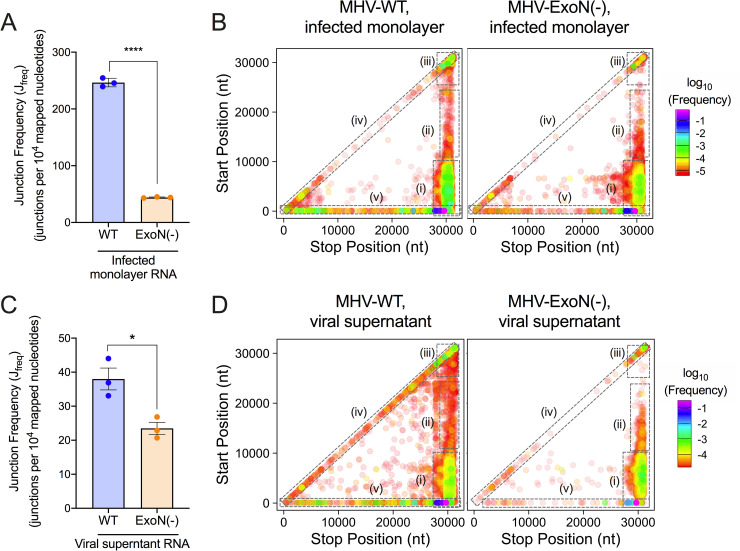
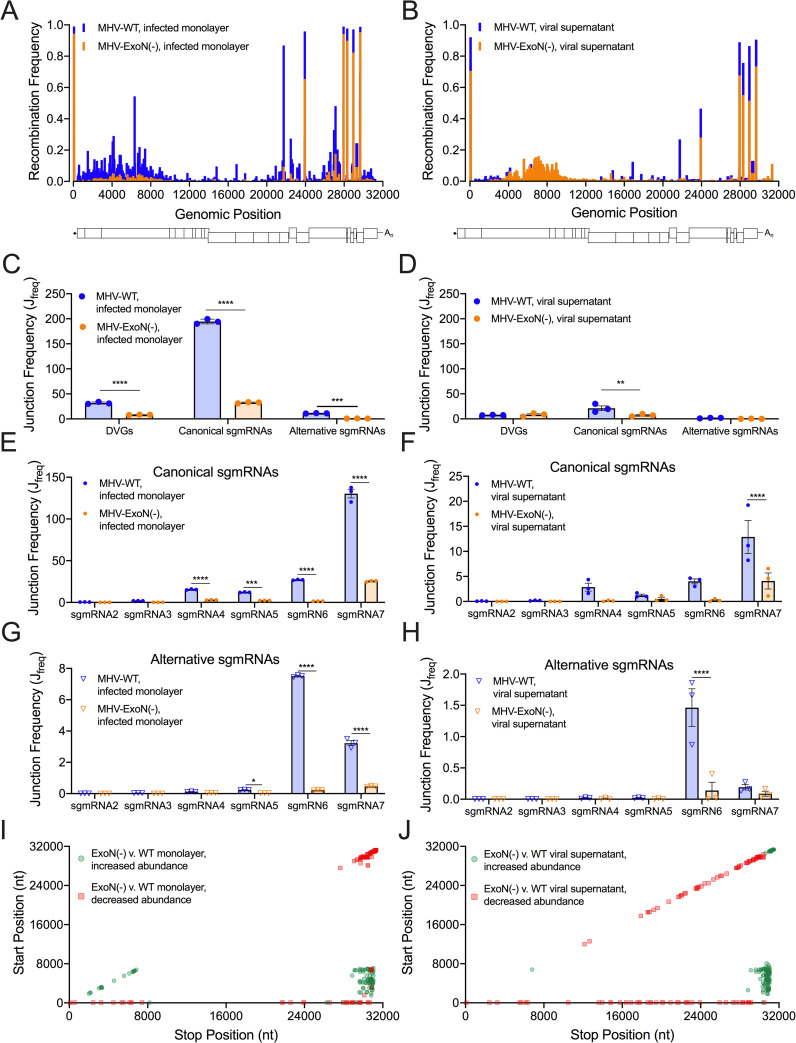
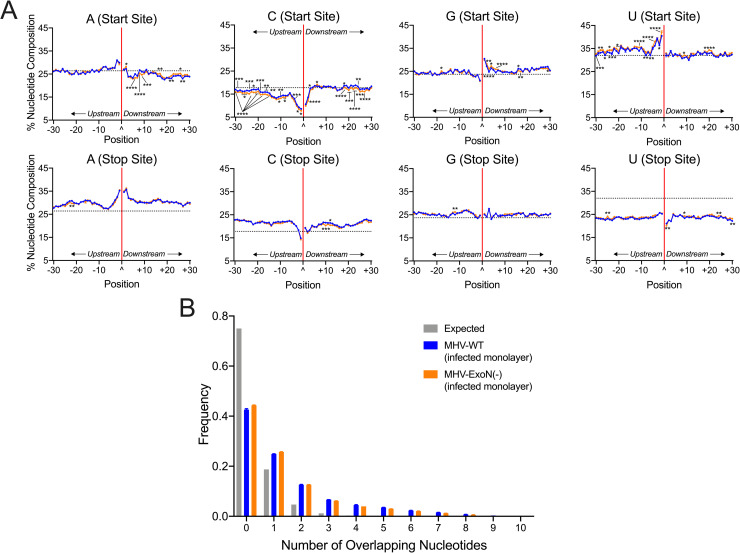
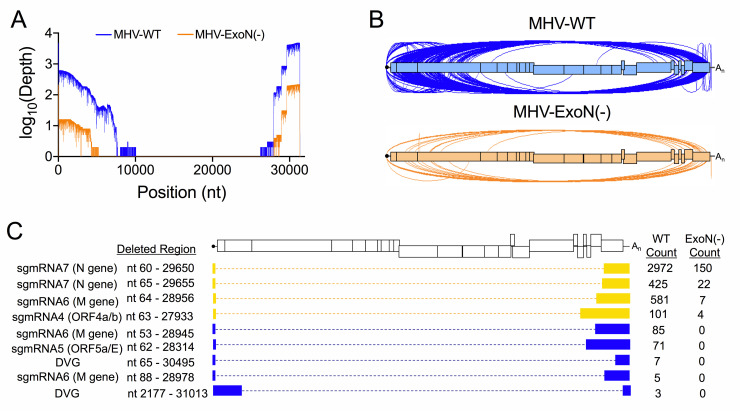
References
-
- Huang J-M, Jan SS, Wei X, Wan Y, Ouyang S. Evidence of the Recombinant Origin and Ongoing Mutations in Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). bioRxiv. 2020. March 19;2020.03.16.993816.
-
- Yi H. 2019 novel coronavirus is undergoing active recombination. Clin Infect Dis [Internet]. 2020. [cited 2020 Mar 11]; Available from: https://academic.oup.com/cid/advance-article/doi/10.1093/cid/ciaa219/578... - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
